Evidence-Based Guideline: Evaluation, Diagnosis, and Mgmt of Facioscapulohumeral Muscular Dystrophy
Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic
Medicine Rabi Tawil, MD, FAAN1; John T. Kissel, MD, FAAN2; Chad Heatwole, MD, MS-CI3; Shree Pandya, PT, DPT, MS4; Gary Gronseth, MD, FAAN5; Michael Benatar, MBChB, DPhil, FAAN6
- MDA Neuromuscular Disease Clinic, School of Medicine and Dentistry, University of Rochester Medical Center, Rochester, NY
- Department of Neurology, Wexner Medical Center, Ohio State University, Columbus, OH
- Department of Neurology, School of Medicine and Dentistry, University of Rochester Medical Center, Rochester, NY
- Department of Neurology, School of Medicine and Dentistry, University of Rochester Medical Center, Rochester, NY
- Department of Neurology, University of Kansas School of Medicine, Kansas City, KS
- Department of Neurology, Miller School of Medicine, University of Miami, Miami, FL
ABSTRACT
Objective: To develop recommendations for the evaluation, diagnosis, prognostication, and treatment of facioscapulohumeral muscular dystrophy (FSHD) from a systematic review and analysis of the evidence.
Methods:
Relevant articles were analyzed in accordance with the American Academy of Neurology classification of evidence schemes for diagnostic, prognostic, and treatment studies. Recommendations were linked to the strength of the evidence and other factors.
Results and recommendations: Available genetic testing for FSHD type 1 is highly sensitive and specific. Although respiratory insufficiency occurs rarely in FSHD, patients with severe FSHD should have routine pulmonary function
testing. Routine cardiac screening is not necessary in patients with FSHD without cardiac symptoms. Symptomatic retinal vascular disease is very rare in FSHD. Exudative retinopathy, however, is potentially preventable, and patients with large deletions
should be screened through dilated indirect ophthalmoscopy. The prevalence of clinically relevant hearing loss is not clear. In clinical practice, patients with childhood-onset FSHD may have significant hearing loss. Because undetected hearing loss
may impair language development, screening through audiometry is recommended for such patients. Musculoskeletal pain is common in FSHD, and treating physicians should routinely inquire about pain. There is at present no effective pharmacologic intervention
in FSHD. Available studies suggest that scapular fixation is safe and effective. However, these studies used different surgical approaches and rarely defined patient selection criteria. Surgical scapular fixation might be cautiously offered to selected
patients. Aerobic exercise in FSHD appears to be safe and potentially beneficial. On the basis of the evidence, patients with FSHD might be encouraged to engage in low-intensity aerobic exercises.
INTRODUCTION
Facioscapulohumeral muscular dystrophy (FSHD) is the third most common form of muscular dystrophy (MD), with a prevalence of approximately 1:15,000–1:20,000.e1,e2 It is an autosomal dominant disorder; however, up to 30% of cases are sporadic, arising from de novo mutations. FSHD is characterized by a distinctive, initially regional distribution of muscle involvement. As the name implies, facial, periscapular, and humeral muscles typically are involved early in the disease course, although the deltoids are spared. This regional involvement, often asymmetric, leads to a distinctive appearance to the shoulders of straight clavicles and scapular winging on attempted shoulder abduction or forward flexion.e3
FSHD symptoms typically develop in the second decade of life but can begin at any age from infancy to late adulthood. As many as one-third of patients are asymptomatic, with the diagnosis made on the basis of previously unrecognized physical examination signs, present in more than 90% of patients by the age of 20.e1
Shoulder girdle weakness, often asymmetric, is the most common presenting symptom. Weakness progresses in a descending manner to involve the upper arm muscles, then the trunk and abdomen, and then the lower extremities, especially the ankle dorsiflexors. FSHD typically progresses slowly but variably.e4,e5 About 20% of individuals with FSHD become wheelchair dependent after age 50.e1 Clinically relevant extramuscular manifestations are uncommon in FSHD but can include respiratory compromise; retinal vascular disease that, in rare cases, leads to an exudative retinopathy and visual loss; hearing loss; and, possibly, an increased incidence of cardiac arrhythmias.
The molecular genetic basis of FSHD is complex. At the tip of chromosome 4q35 lies a repetitive 3.3 kb DNA sequence known as D4Z4 repeats.e6,e7 Moreover, there are 2 different DNA variants distal to the D4Z4 repeats, called the A and B allelic variants.e8 FSHD type 1 (FSHD1), accounting for 95% of FSHD cases, results from deletion of a critical number of D4Z4 repeats, but only when this occurs on the A allele. The biological basis for this dual requirement is becoming increasingly understood. Contraction of the D4Z4 repeat results in a more open chromatin structure, allowing the potential expression of gene sequences within the repeats. One such gene, double homeobox 4 (DUX4), lacks the polyadenylation (poly-A) sequence required to produce stable messenger RNA.e9,e10 Because only the A (not the B) allele variant contains a poly-A sequence, stable DUX4 expression can occur only in the presence of the A allelic variant.e11,e12
Complicating matters is the existence of a genetically distinct but clinically identical FSHD type—FSHD type 2 (FSHD2)—now known to account for approximately 5% of patients with clinically defined FSHD.e13,e14 Unlike the majority of patients with FSHD (i.e., FSHD1), patients with FSHD2 do not have contractions in the 4q35 D4Z4. As with FSHD1, and despite a normal number of repeats, the chromatin structure at the D4Z4 repeats is more open, and at least one 4q35 allele is an A variant.e13 Recent studies have implicated mutations in SMCHD1, a gene on chromosome 18 that functions as a chromatin modifier, as the cause of the D4Z4 chromatin changes observed in about 85% of patients with FSHD2.e15 Comprehensive molecular genetic testing for FSHD2 is complex and not readily available currently, and thus is not addressed in this guideline.
Despite having distinct genotypes, FSHD1 and FSHD2 have an identical molecular basis that results from the aberrant expression of the DUX4 gene in skeletal muscle.e15,e16 DUX4 protein is a transcription factor normally expressed only in the germline, but little is known about its function.e17 Preliminary evidence suggests that inappropriate expression of DUX4 and its transcriptional targets in skeletal muscle can result in apoptosis, impaired muscle regeneration, and induction of an immune response.e17
The clinical diagnosis of FSHD is based on the presence of a characteristic distribution of muscle weakness and is easily confirmed in most instances of FSHD1 by genetic testing. To date there is no effective treatment for muscle weakness in FSHD. Standard disease management includes physical therapy, bracing for foot drop, surgical scapular fixation in some patients, management of respiratory complications, and management and symptomatic treatment of extramuscular manifestations.
Previous FSHD practice guidelines have been based on consensus and expert opinion.e18,e19 The present guideline, based on systematic review of the evidence, focuses exclusively on FSHD. Duchenne MD and myotonic dystrophy will be discussed in forthcoming guidelines; limb-girdle muscular dystrophy and congenital MD are addressed in separate guidelines.e20,e21 The present guideline addresses the following practical issues related to FSHD (reflective only of evidence relevant to FSHD1; no large FSHD2 clinical studies exist):
- In regard to genetic testing, for patients with clinically defined FSHD (as determined by explicitly stated clinical criteria substantially similar to the consortium criteria),e22 how often does D4Z4 contraction on 4q35 confirm the diagnosis of FSHD (irrespective of its occurrence on an allele A background)? For individuals who do not have FSHD, how often is a D4Z4 contraction on 4q35 found? For individuals who do not have FSHD, how often is a D4Z4 contraction on 4q35 on allele A found?
- Among patients with FSHD, which factors are associated with or predict loss of clinically meaningful milestones (e.g., loss of independent ambulation)?
- Among patients with FSHD, how frequent are respiratory abnormalities, cardiac abnormalities, retinal disease, hearing loss, and pain?
- In regard to treatment, do interventions (as compared with no intervention) improve patient- relevant outcomes? Are there features that identify patients who are more or less likely to improve with a specific intervention?
DESCRIPTION OF THE ANALYTIC PROCESS
In July 2010, the Guideline Development, Dissemination, and Implementation Subcommittee (GDDI) of the American Academy of Neurology (AAN) and the Practice Issues Review Panel (PIRP) of the American Association of the Neuromuscular & Electrodiagnostic
Medicine (AANEM) convened a panel of clinicians with expertise in FSHD (see appendices e-1 and e-2 for a listing of the members of the AAN GDDI and AANEM PIRP). In accordance with the processes outlined in the 2004 and 2011 AAN guideline development
manuals,
e23,e24 the panel searched the Medline, EMBASE, Cochrane, and Scopus databases from 1948 to October 2012 for relevant peer-reviewed articles in humans and in all languages (see appendix e-3 for search strategies). The initial search yielded
977 abstracts. Of those, 176 were obtained for full-text review. Each of the 176 articles was reviewed by 2 panel members working independently of each other. A total of 94 articles were selected for inclusion in the analysis, and of those, 76 articles
were selected for evidence rating. An updated literature search completed in January 2014 identified an additional 12 potentially relevant articles, 4 of which were selected for evidence rating.
Selected articles contained information relevant
to the 4 questions posed above and had acceptable study designs, including randomized, controlled trials; cohort studies; case-control
studies; and case series. Reviews and meta-analyses were excluded, as were studies with 6 or fewer participants
for studies of FSHD complications and prognosis, fewer than 9 participants
for genetic screening, and fewer than 5 participants for treatment. Also excluded were studies not relevant to the clinical questions, studies including participants who
had unrelated diseases or were outside of the study population, and articles that were not peer reviewed. Each of the 76 articles was rated by 2 panel members using the AAN criteria for classification of screening, prognostic, and treatment articles
(appendix e-4).
The panel formulated a rationale for recommendations based on the evidence systematically reviewed and stipulated axiomatic principles of care. This rationale is explained in a section that precedes each set of recommendations.
From this rationale, corresponding actionable recommendations were inferred. The level of obligation of the recommendations was assigned using a modified Delphi process that considered the following prespecified domains: the confidence in the evidence
systematically reviewed, the acceptability of axiomatic principles of care, the strength of indirect evidence, and the relative magnitude of benefit to harm. Additional factors explicitly considered by the panel that could modify the level of obligation
include judgments regarding the importance of outcomes, cost of compliance with the recommendation relative to benefit, the availability of the intervention, and anticipated variations in patients’ preferences. The prespecified rules for determining
the final level of obligation from these domains are indicated in appendix e-5. The level of obligation was indicated using standard modal operators. Must corresponds to Level A, very strong recommendations; should to Level B, strong recommendations;
and might to Level C, weak recommendations. The panel members’ judgments supporting the levels of obligation are indicated in appendix e-6.
ANALYSIS OF EVIDENCE
FSHD genetic testing.
Clinical questions.
Understanding the molecular genetics of FSHD1 is critical to the molecular diagnosis of this disorder. Healthy individuals possess at least 11 D4Z4 repeats, yielding a DNA fragment >38 kb on standard genetic testing. Affected individuals, in contrast, possess between 1 and 10 repeats, yielding DNA fragments 10 to 38 kb in size.e7 Measurement of the size of the residual D4Z4 sequence on 4q35 forms the basis for genetic testing in FSHD. As previously discussed, an additional requirement for FSHD identification is that the contraction occur on the A allelic variant. Routine first-pass commercial genetic testing in the United States measures the residual D4Z4 repeat sizes without determining the A or B allelic variants. The prevalence of D4Z4 repeat sizes in the range of 1 to 10 alleles is low in the general population. This low prevalence raises questions about the clinical utility of routine determination of the A/B variant in molecular confirmation of FSHD. The following specific clinical questions were examined:
- For patients with clinically defined FSHD (as determined by explicitly stated clinical criteria substantially similar to the consortium criteria), how often does D4Z4 contraction on 4q35 confirm the diagnosis of FSHD (irrespective of its occurrence on an allele A background)?
- For individuals who do not have FSHD, how often is a D4Z4 contraction on 4q35 found?
- For individuals who do not have FSHD, how often is a D4Z4 contraction on 4q35 on allele A found?
Analysis.
Nine Class III studies containing information relevant to the questions above were reviewed.e25– e33 The studies were rated Class III primarily because the patient populations studied were recruited from specialty clinics, which increases
the risk of referral bias.
All 9 Class III studies addressed the question of the sensitivity of the D4Z4 contraction on 4q35 for the diagnosis of FSHD.e25–e33 In these studies FSHD was defined by standard clinical criteria.
In most of the studies the deletion was detected by measurement of allele size through use of the standard p13E-11 probe proximal to the D4Z4 repeat and genomic DNA digested with EcoRI and BlnI restriction enzymes. The frequency of D4Z4 contractions
among patients with clinically defined FSHD ranged from 86% to 100%. There was statistical heterogeneity in the results (I2 = 0.65). Some of the heterogeneity in the studies could be accounted for by the varying definitions for the upper limit in
the size of a deleted allele (range: 28–38 kb). The pooled estimate of the sensitivity of the presence of D4Z4 contraction (random effects) was 93% (95% confidence interval [CI] 88%–96%). The confidence in the evidence was graded as moderate
(upgraded from low because of the magnitude of the effect).
These 9 studies also contained evidence in regard to the specificity of the presence of the D4Z4 contraction.e25–e33 One of the studies was excluded because
the genetic testing was done using a single digestion with EcoRI, which could result in false-positive contraction on the homologous region on chromosome 10.e28 The frequency of D4Z4 contractions in normal controls ranged from 0% to 9%.
The pooled estimate of the specificity of the presence of D4Z4 contraction (random effects) was 99% (95% CI 97%–100%). The confidence in the evidence was graded as moderate (upgraded from low because of the magnitude of the effect).
A single Class III study contained evidence in regard to the specificity of the presence of a D4Z4 contraction on an A allele background.e29 This study found that 11 out of 801 normal individuals carried a contracted allele on an
A allele background (specificity 98%; 95% CI 97.5%–99.2%). The confidence in the evidence was graded as low (upgraded from very low because of the magnitude of the effect).
Conclusions.
The finding of a D4Z4 contraction on chromosome 4q35 likely has a sensitivity of 93% and a specificity of 98% for the diagnosis of clinically defined FSHD (9 Class III studies).e25–e33 In a patient population with clinically defined FSHD, the degree
of specificity is not likely to be further enhanced by testing for presence of the A variant.
Risk factors for disease severity.
Clinical question.
A critical aspect of management of patients with any neuromuscular disorder lies in identifying clinical, biochemical, or genetic aspects of the illness associated with prognosis. It is indispensable to identify such risk factors that might be linked
to a severe (or more benign) course when discussing prognosis with patients, designing therapy programs and other meaningful interventions, and helping patients make important medical, financial, and other life decisions. This is true particularly
in a disease such as FSHD where there is tremendous variability in the extent and severity of involvement. For this analysis, relevant studies were reviewed to address the following specific question: Among patients with FSHD, which factors are associated
with or predict loss of clinically meaningful milestones (e.g., loss of independent ambulation)?
Analysis.
Seven studies containing information relevant to this question were reviewed.e30,e34–e39 One study was performed before accurate genetic testing was available and was not considered further.e36 The remaining studies explored
the prognostic effects of 2 risk factors: age at symptom onset and D4Z4 repeat size. Five studies examined the relationship between D4Z4 repeat size and severity.e30,e34,e35,e38,e39 A Class I study in a cohort of 313 patients showed a linear
relationship between age at diagnosis and repeat size. The study also showed that the age at which patients started using wheelchairs is associated with D4Z4 repeat size: 24.1 years (CI 17.0–31.3) for repeat sizes of <18 kb, 48 years (CI
44.0–52.3) for repeat sizes of 19 to 28 kb, and 58.6 years (CI 52.2–64.9) for repeat sizes of > 28 kb.e38 A Class II study found a similar correlation between age at loss of ambulation and repeat size (r = 0.773, p <
0.001).
e39 Another Class II study in a cohort of 165 patients with FSHD found an inverse correlation between fragment size and clinical severity as assessed by degree of leg weakness and a global clinical severity score.e30 Severe lower-limb
involvement was found in 100% of patients with an EcoRI fragment size of 10 to 13 kb, in 53% of patients with a fragment size of 16 to 20 kb, and in only 19% of patients with a fragment size larger than 21 kb. In this study 36% of the variation
in the severity of lower-limb involvement was explained by fragment size. There was no significant correlation found between fragment size and age at loss of ambulation. In a Class III study of 7 de novo patients, quantitative isometric myometry test
(QMT) scores normalized for age, sex, and height were used to quantify overall disease severity. This analysis found a significant (r = 0.92, p < 0.004) correlation between disease severity and the size of the 4q35-associated deletion.
e36 A Class II study of 65 patients, however, found no correlation between Clinical Severity Scale scores and fragment size or between fragment size and physical function on the SF-36 quality of life (QOL) scale; however, on the physical
function subscore of this scale, patients with fragment sizes <18 kb had lower scores (p value not reported).e37
A single Class III study addressed the question of whether age at onset affected disease severity.e34 This study found a significant correlation between proband age at onset and FSHD- associated fragment size (r = 0.56, p < 0.001). A similar correlation (r = 0.70, p < 0.01) with fragment size was observed for age at loss of ambulation in 16
patients using a wheelchair.
Conclusions.
In patients with FSHD, smaller D4Z4 repeat size is probably associated with more severe disease as measured by age at diagnosis and age at wheelchair dependence (1 Class I study).e38 Other measures of disease severity possibly associated with
smaller fragment size include quantitative computerized muscle testing, severity of leg weakness, global severity scores, and earlier loss of ambulation (one Class II study or multiple Class III studies).e30,e34,e35,e39 Earlier age at onset
is also possibly associated with smaller fragment size and earlier loss of ambulation (one Class III study).e34 Patients with very large deletions (EcoRI fragment sizes of 10–15 kb) are particularly prone to severe disease.
Complications.
Clinical question.
Although the cardinal features of FSHD involve limb weakness that starts with focal weakness of the shoulders, face, and humeral muscles, additional systemic features may occur. These extramuscular features may have significant and, at times, life-threatening
consequences. It is important for clinicians to recognize the association between these manifestations and the presence of FSHD so that needed monitoring, counseling, and early interventions may be implemented. The following specific clinical
question was examined: Among patients with FSHD, how frequent are respiratory abnormalities, cardiac abnormalities, retinal disease, hearing loss, and pain?
Analysis.
Respiratory abnormalities. One Class II study, 1 Class III study, and 1 Class IV study were analyzed.e36,e40,e41 In the Class II study, 10 patients with FSHD with respiratory insufficiency requiring nocturnal ventilator support were identified
in a Dutch FSHD population of 800 patients, representing an estimated prevalence of 1.25% (95% CI 0.5%–2%).e40 The Class III study examined pulmonary function testing (PFT) in 23 of 53 patients with FSHD.e36 All patients
had clinically defined FSHD, but it was not genetically confirmed. Of the 23 patients tested, 3 or 13% (95% CI 0.7%–27%) had a severe restrictive pattern on PFT. In contrast, the Class IV study selected 16 patients with genetically confirmed
FSHD who were ambulant but severely affected and found only mild signs of a restrictive pattern on PFT in some patients (forced vital capacity [FVC] range 85%–117% predicted).e41
Cardiac abnormalities. Four Class
III studies using electrocardiography/echocardiography found no structural abnormalities in 80 patients with FSHD (95% CI 0%–4.6%).e42–e45 Six Class III studies examined surface electrocardiogram/echocardiogram in a combined
total of 227 patients with FSHD.e36,e42-e46 Abnormalities were found in 89 or 39.2% (95% CI 33.1%–45.7%) of patients screened. The same 6 Class III studies looked at the frequency of symptomatic or inducible supraventricular arrhythmias
and found these in 22 patients or 9.7% (95% CI 6.5%– 14.2%).
Four Class III studies examined the frequency of retinal vascular abnormalities on dilated eye examination or fluorescein angiography in 294 patients.e47–e50 Of those screened, 15 or 25% (95% CI 20.9%–30.8%) had retinal vascular abnormalities. One of the Class III studies, examining 396 patients with genetically confirmed FSHD, identified 3 patients with symptomatic retinal vascular disease.e50 For the 4 studies, the combined proportion of patients with FSHD who had symptomatic retinal disease is 0.6% (95% CI 0.2%–1.5%).e47–e50 In the previously mentioned study, a patient survey and literature review of patients with
FSHD who had Coats disease, a total of 14 patients were identified; all but one had very large deletions (< 20 kb).e50
Hearing loss. Eight Class III studies used audiometry to examine hearing in a combined total of 394
patients. Of the patients examined, 61 or 15.5% (95% CI 12.1%–19.4%) had audiometric abnormalities.e26,e33,e47–e49,e51–e53 In one of the studies, 3 of 4 patients followed with sequential audiometry over a 5-year period
showed worsening hearing loss by audiogram.e53 In addition, as in symptomatic retinal vascular disease, hearing loss occurs only in patients with large deletions (<20 kb); hearing loss occurs in 32% (95% CI 16.7%–51.4%) of patients
with large deletions.e53 This observation is supported by a study of patients with FSHD and both symptomatic retinal vasculopathy and large deletion size.e50 Of the 14 patients identified, 57% (95% CI 29.0%–
82.3%) had hearing loss, 35.7% of whom required hearing aids.e50
Pain. One Class II study and 2 Class III studies examined the frequency of pain in a combined total of 376 patients with FSHD.e37,e54,e55 Of those surveyed,
297 or 79% (95% CI 74.6%–82.8%) complained of pain. A single study assessed the severity of pain. Of 65 patients in that study, 8 or 10.8% (95% CI 3.2%–18.3%) had clinically significant pain.e37 The most common sites of
pain are, in descending order, the lower back, the legs, the shoulders, and the neck.e54
Conclusions.
Respiratory abnormalities. Evidence suggests that respiratory insufficiency and reduced pulmonary function may occur. However, there is insufficient evidence to determine the frequency and severity of respiratory compromise in patients with FSHD (1 Class
II studye40 and 1 Class III studye36).
Cardiac abnormalities. The prevalence of structural cardiac abnormalities on electrocardiography/echocardiography is possibly zero, but precision of this estimate cannot exclude a frequency of up to
4.6%. Although symptomatic or inducible supraventricular arrhythmias are found in patients with FSHD, because of the risk of referral bias there is insufficient evidence to determine the frequency of clinically relevant supraventricular arrhythmias
(multiple Class III studies).e36,e42–e46
Retinal vascular disease. Confidence in the evidence for the prevalence of retinal vascular abnormalities is low, with up to 25% (95% CI 20.9%–30.8%) showing abnormalities
on examination. However, only 0.6% (95% CI 0.2%–1.5%) of patients with FSHD develop symptomatic retinal disease (4 Class III studies).e47–e50
Hearing loss. Confidence in the evidence for the prevalence of audiometric
abnormalities is very low due to the wide range of prevalence reported, ranging from rates equivalent to a normal matched control population to a prevalence of 64%. One study that correlated hearing loss with genotype suggests that only patients with
large deletions, who represent about 15% of all patients with FSHD, are susceptible to hearing loss.e53 Poor representation of this subgroup in some studies could account for the wide range of prevalence.
Pain. There is a high
prevalence of pain in patients with FSHD, likely up to 79%, with a low level of confidence in the evidence (1 Class II study and 2 Class III studies).e37,e54,e55 However, the prevalence of clinically significant pain, as reported in
a single Class III study, is likely much lower at 10.8%.e37
Treatment.
Clinical questions.
The goal of therapy in FSHD is to improve muscle strength or function, or both. Until recently the underlying pathophysiology of FSHD was unknown, and thus pharmacologic trials have focused on improving muscle mass and strength, whereas surgical studies of scapular fixation have been motivated by efforts to improve function notwithstanding the presence of weakness. The trial of albuterol, for example, was based on animal studies showing an anabolic effect of the similar β2-agonist drug clenbuterol. Likewise, the discovery of myostatin as an inhibitor of muscle growth has generated an interest in the use of myostatin inhibitors (such as MYO-029) in the hope that these would lead to increased muscle mass and strength. In the absence of effective pharmacologic therapy, a number of strategies have been used to address the problem of weakness of scapular stabilizers, as scapular destabilization is the most prominent manifestation of the disease, affecting more than 90% of individuals.e38 This has proven stubbornly refractory to effective bracing, providing a motivation for surgical fixation in carefully selected patients. Relevant studies that were reviewed include studies on pharmacologic interventions, exercise, and surgical scapular fixation, or a combination of these modalities. The following questions were examined:
- Do interventions (as compared with no intervention) improve patient-relevant outcomes?
- Are there features that identify patients who are more or less likely to improve with a specific intervention?
Analysis.
Pharmacologic interventions. Three Class I studies of pharmacologic interventions provided pertinent information. Two randomized, controlled trials (Class I) examined the effect of oral albuterol on strength in FSHD, in one study after 12 months of treatment and in the second study after 6 months of treatment.e56,e57 One study showed no effect on a global measure of strength based on QMT.e57 The other study showed a clinically unimportant increase in the strength of 7 of 12 muscles measured by QMT.e56 A third Class I randomized, controlled study examined the effect of an IV myostatin inhibitor (MYO-029) and showed no significant improvement in muscle strength.e58 Three additional studies of pharmacologic intervention in FSHD were excluded because all were graded Class IV. One study examined the effect of prednisone on strength, a second study examined the effect of diltiazem on strength, and a third study examined the effect of albuterol on pain and fatigue. e59,e60,e61 All three studies showed no beneficial effect.
Surgical scapular fixation. Eleven studies were reviewed on scapular fixation surgery; one was Class III and 10 were Class IV.e62—e72All the studies were uncontrolled case series and involved different surgical approaches. The consistency of the response on parameters of shoulder function (forward flexion, abduction, and pain) resulted in the upgrading of the confidence in the studies from very low to low.
Exercise. Two studies provided relevant information on the role of exercise. One 52-week Class I study examined the effect of strength training on muscle strength and showed no evidence of improved isometric strength testing; however, it showed improvement
of significant but questionable importance in dynamic strength in 1 of 2 muscle groups tested.e59 A single Class III study of 8 patients with FSHD showed that 12 weeks of low-intensity aerobic exercise improved VO2 max by 16% (standard
deviation [SD] 3; p < 0.002) and workload by 17% (SD 4; p < 0.002), as well as self-reported levels of activity, without evidence of muscle damage.e73
Conclusions.
Pharmacologic interventions. It is highly likely that albuterol is ineffective for improving muscle strength (2 Class I studies).e56,e57 However, there is insufficient evidence to judge the efficacy of albuterol for muscle pain and fatigue
(single Class IV study).e59 The myostatin inhibitor MYO- 029 is probably ineffective for improving muscle strength, pulmonary function, timed function, and QOL (single Class I study).e58 There is insufficient evidence to support
or refute the effects of prednisone (single Class IV study) or diltiazem (1 Class IV study) on muscle strength.e60,e61
Surgical scapular fixation. Scapular fixation is possibly effective for improving shoulder abduction and anterior flexion (1 Class III study, 9 Class IV studies) as well as shoulder pain (2 Class IV studies).e62–e72 (The confidence in the evidence for scapular fixation was upgraded from very low to low because of the magnitude of effect.)
Exercise. On the basis of a single Class I study, strength-training exercise is probably ineffective for improving muscle strength meaningfully (1 Class I study).e59 There is evidence that supports the use of aerobic exercise in FSHD, but the
confidence in the evidence, based on a single Class III study, is very low.e73
PRACTICE RECOMMENDATIONS
The recommendations below encompass four major areas: diagnosis, predictors of severity, surveillance for complications, and treatment of FSHD. Each recommendation is preceded by a clinical context section that outlines the evidence, general principles
of care, and evidence from related disorders that inform the recommendations.
Because of the relative paucity of literature directly relevant to FSHD, for some of the clinical questions, some of the recommendations below are based in part
on evidence from other neuromuscular disorders.
Diagnosis of FSHD.
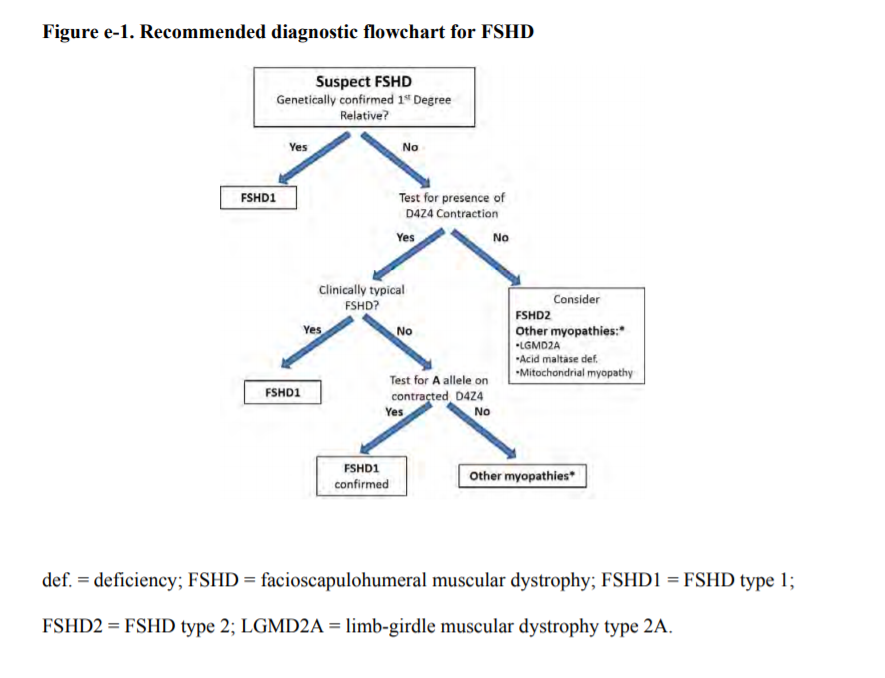
See also the algorithm in figure e-1.
Clinical context.
When clinical presentation of FSHD is typical and the inheritance pattern is consistent with autosomal dominant inheritance, clinical diagnosis is usually straightforward. If, in such circumstances, the diagnosis is genetically confirmed in a first-degree
relative, genetic testing is not necessary for each affected individual. However, atypical presentations are not uncommon. In the setting of atypical or sporadic cases, genetic confirmation is important for genetic counseling, especially with the
recent discovery of 2 genetically distinct forms of FSHD (PRIN). In the most common FSHD type, FSHD1, disease results from contraction of a DNA repeat sequence, termed D4Z4 repeat, on one copy of 4q35 from more than 10 repeats to 1 to 10 repeats.
In addition, the contraction must occur in the presence of one particular (A variant) of 2 (A/B) sequence variants distal to the repeats (PRIN). Available molecular testing for FSHD1, which measures only the presence of a repeat contraction on initial
testing, is highly sensitive and specific (EVID). In studies that utilized strict diagnostic criteria for FSHD, determining whether a contraction occurs on an A variant genetic background does not appear to improve diagnostic specificity (EVID). However,
in clinical practice, strict clinical diagnostic criteria might not be adhered to, increasing the chances of a false-positive result (INFER). In consequence, determining that a D4Z4 contraction is occurring on an A variant is warranted
when the clinical presentation is atypical for FSHD. At present, commercial genetic testing in FSHD is limited to FSHD1 testing.
Recommendation.
| A1. | Clinicians should obtain genetic confirmation of FSHD1 in patients with atypical presentations and no first-degree relatives with genetic confirmation of the disease (Level B). Figure e-1 shows the recommended FSHD molecular diagnosis decision tree. |
Predictors of severity in FSHD.
Clinical context.
Factors that predict disease severity in FSHD are important for counseling patients and for screening for and managing potential complications (PRIN). The D4Z4 deletion size appears to be somewhat predictive of the overall rate of disease progression
(EVID). D4Z4 deletion size should be used cautiously for predicting disease progression rate in any particular individual due to other sources of variation affecting disease severity, including intrafamilial factors (INFER). Clinical experience suggests
that patients with severe childhood-onset disease almost invariably have very large deletions (i.e., contracted D4Z4 allele of 10–20 kb or 1–4 repeats), suggesting a much more robust correlation between disease severity and large deletions
(EVID).
Recommendation.
| B1. | Large D4Z4 deletion sizes (contracted D4Z4 allele of 10–20 kb) should alert the clinician that the patient is more likely to develop more significant disability and at an earlier age. Patients with large deletions are also more likely to develop symptomatic extramuscular manifestations (Level B). (See next section on monitoring for FSHD complications). |
Monitoring for complications of FSHD.
Pulmonary complications.
Clinical context.
Our systematic review revealed that some patients with FSHD develop respiratory muscle weakness that can result in respiratory failure and need for mechanical ventilator assistance (e.g., nocturnal bilevel positive airway pressure), although this complication
is uncommon (EVID). Patients with chronic respiratory failure from neuromuscular-related weakness often do not have classic symptoms of ventilatory failure (i.e., overt dyspnea). Impending respiratory failure, therefore, may begin with respiratory
insufficiency mainly during sleep, resulting in excessive daytime somnolence or nonrestorative sleep. Respiratory insufficiency in patients with FSHD, therefore, may be evident only through pulmonary function testing (PRIN). Respiratory failure constitutes
a major source of morbidity in patients with most MD types and can severely disrupt sleeping, daily activities, and QOL (PRIN). Early intervention with noninvasive mechanical ventilation leads to improved survival and QOL (RELA).e74
Recommendations.
| C1. | Clinicians should obtain baseline pulmonary function tests on all patients with FSHD. Patients should be monitored regularly if they have abnormal baseline pulmonary function test results or any combination of severe proximal weakness, kyphoscoliosis, wheelchair dependence, or comorbid conditions that may affect ventilation (e.g., chronic obstructive pulmonary disease, cardiac disease) (Level B). |
| C2. | In patients who have FSHD and either 1) compromised pulmonary function studies (e.g., FVC <60%) or 2) symptoms of excessive daytime somnolence or nonrestorative sleep (e.g., frequent nocturnal arousals, morning headaches), clinicians should refer patients for pulmonary or sleep medicine consultation for consideration of nocturnal sleep monitoring or nocturnal noninvasive ventilation in order to improve QOL (Level B). |
| C3. | Patients with FSHD who do not get regular pulmonary function testing should be tested prior to surgical procedures requiring general anesthesia, as such testing may uncover asymptomatic respiratory compromise (Level B). |
Cardiac abnormalities.
Clinical context.
Our systematic review revealed very little evidence for structural cardiac abnormalities in FSHD. Also, data are insufficient to suggest that patients with FSHD are susceptible to cardiac arrhythmias (EVID). Routine electrocardiographic or echocardiographic
testing is therefore unnecessary in patients with FSHD who are asymptomatic (INFER).
Recommendation.
| C4. | Patients with FSHD should be referred for cardiac evaluation if they develop overt signs or symptoms of cardiac disease (e.g., shortness of breath, chest pain, palpitations). However, routine cardiac screening is not essential in the absence of cardiac signs or symptoms (Level C). |
Retinal vascular disease.
Clinical context.
Our systematic review suggests that symptomatic retinal vascular disease in the form of an exudative retinopathy (Coats disease) is very rare in FSHD but tends to affect patients with large deletions almost exclusively (EVID). Untreated exudative
retinopathy can lead to significant visual loss, which may be prevented by early intervention (INFER).
Recommendation.
| C5. | Clinicians should refer patients with FSHD and large deletions (contracted D4Z4 allele of 10–20 kb) to an experienced ophthalmologist (e.g., retina specialist) for dilated indirect ophthalmoscopy (Level B). The presence and severity of retinal vascular disease at initial screening should be used to determine the frequency of subsequent monitoring (Level B). |
Hearing loss.
Clinical context.
Our systematic review shows that the available studies fail to capture the prevalence and clinical relevance of hearing loss in FSHD (EVID). In clinical practice, most patients with FSHD and hearing loss requiring the use of a hearing aid have childhood-onset
FSHD with large D4Z4 deletions. Two recent studies support this clinical impression (EVID). Moreover, one of the studies suggests that hearing loss is progressive in some patients. Adults and older children are cognizant of the hearing loss onset,
and therefore intervention can occur early when required. However, failure to detect hearing loss in infants and younger children may significantly delay or impair language development (PRIN).
Recommendation.
| C6. | Clinicians should screen all young children with FSHD at diagnosis and yearly thereafter until these children start school, as hearing loss may not be present at diagnosis and can be progressive (Level B). |
Pain.
Clinical context.
Pain is a common complaint in FSHD and appears to be mostly musculoskeletal in origin (EVID). Pain compounding muscle weakness can have a significant impact on QOL (INFER). Physical therapists often can provide insight into the mechanism of pain in patients
with weakness (PRIN). Nonsteroidal anti-inflammatory medications are useful for acute pain, and antidepressants or antiepileptics, for chronic musculoskeletal pain (PRIN).
Recommendation.
| C7. | Treating physicians should routinely inquire about pain in patients with FSHD. Referral for a physical therapy evaluation may prove helpful as an initial nonpharmacologic intervention. In patients with persistent pain and no contraindications, a trial of nonsteroidal anti-inflammatory medications is appropriate for acute pain and antidepressants or antiepileptics for chronic pain (Level B). |
Treatment of FSHD.
Pharmacologic interventions.
Clinical context.
As of this writing, no evidence exists for any effective pharmacologic interventions that improve strength or slow disease progression in FSHD. Randomized, controlled trials of albuterol were negative (EVID). Uncontrolled, open-label trials of corticosteroid
and diltiazem showed no benefit. A controlled early phase II study of MYO-029, a myostatin inhibitor, also failed to show benefit.
Recommendation.
| D1. | In patients with FSHD, clinicians should not prescribe albuterol, corticosteroid, or diltiazem for improving strength (Level B). |
Surgical scapular fixation.
Clinical context.
In patients with FSHD, limited shoulder range of motion due to periscapular muscle weakness is a major source of functional limitation (PRIN). Moreover, in many patients, bedside manual scapular fixation can result in significant improvement in shoulder
range of motion (PRIN). Postoperative complications are infrequent but include hemo- or pneumothorax, pain, infection, non-union, and reduced lung capacity. Scapular fixation appears to be generally safe and may be effective for improving shoulder
range of motion (EVID).
Recommendation.
| D2. | Surgical scapular fixation might be offered cautiously to selected patients after careful consideration of the overall muscle impairment in the involved arm, assessment of potential gain in range of motion by manual fixation of the scapula, the patient’s rate of disease progression, and the potential adverse consequences of surgery and prolonged postsurgical bracing (Level C). |
Aerobic exercise.
Clinical context.
Aerobic exercise in FSHD appears to be safe and potentially beneficial (EVID), as has been shown in many other muscle diseases (RELA).e75 Aerobic fitness is important for overall health (PRIN). To minimize injury from falls or overuse, the
type of aerobic exercise should be tailored to the patient’s particular distribution of weakness. For example, a stationary bicycle rather than a treadmill should be recommended for patients with leg weakness (PRIN). Although no data
exist to suggest that strength training is detrimental in FSHD (EVID), further research is needed to determine whether such strength training will result in clinically meaningful long-term functional improvement (INFER).
Recommendations.
| D3. | Clinicians might encourage patients with FSHD to engage in low-intensity aerobic exercise. An experienced physical therapist can help guide development of individualized exercise programs. Clinicians might also use the practical physical activities guidelines for individuals with disabilities, provided by the US Department of Health and Human Services, when counseling patients about aerobic exercise (Level C).e76 |
| D4. | In patients interested in strength training, clinicians may refer patients to physical therapists to establish a safe exercise program using appropriate low/medium weights/resistance that takes into consideration the patients’ physical limitations (Level C). |
RECOMMENDATIONS FOR FUTURE RESEARCH
- Future studies in FSHD should employ a clinical definition of disease based on consensus criteria combined with molecular genetic diagnosis.
- Observational studies (e.g., case-control and cohort) should conform to the Strengthening the Reporting of Observational Studies in Epidemiology criteria for reporting of results, leading to higher confidence in the published evidence.e77
- Patients with childhood-onset FSHD appear to have more severe neuromuscular manifestations as well as more clinically significant extramuscular complications. Understanding what risk factors predispose this subpopulation of patients with FSHD to such extramuscular manifestations is important for developing specific management strategies. A concerted effort is needed to study such patients with clearly defined clinical and genetic parameters.
- Use of patient registries will allow researchers to access more patients and obtain more robust data on risk factors and prognostic factors. Such a registry exists in the United States, and others are being developed elsewhere.
DISCLAIMER
Clinical practice guidelines, practice advisories, systematic reviews and other guidance published by the American Academy of Neurology and its affiliates are assessments of current scientific and clinical information provided as an educational service.
The information: 1) should not be considered inclusive of all proper treatments, methods of care, or as a statement of the standard of care; 2) is not continually updated and may not reflect the most recent evidence (new evidence may emerge between
the time information is developed and when it is published or read); 3) addresses only the question(s) specifically identified; 4) does not mandate any particular course of medical care; and 5) is not intended to substitute for the independent professional
judgment of the treating provider, as the information does not account for individual variation among patients. In all cases, the selected course of action should be considered by the treating provider in the context of treating the individual
patient. Use of the information is voluntary. AAN provides this information on an “as is” basis, and makes no warranty, expressed or implied, regarding the information. AAN specifically disclaims any warranties of merchantability or fitness
for a particular use or purpose. AAN assumes no responsibility for any injury or damage to persons or property arising out of or related to any use of this information or for any errors or omissions.
CONFLICT OF INTEREST
The American Academy of Neurology and American Association of Neuromuscular & Electrodiagnostic Medicine are committed to producing independent, critical, and truthful clinical practice guidelines (CPGs). Significant efforts are made to minimize the
potential for conflicts of interest to influence the recommendations of this CPG. To the extent possible, the AAN and AANEM keep separate those who have a financial stake in the success or failure of the products appraised in the CPGs and the
developers of the guidelines. Conflict of interest forms were obtained from all authors and reviewed by an oversight committee prior to project initiation. AAN and AANEM limit the participation of authors with substantial conflicts of interest.
The AAN and AANEM forbid commercial participation in, or funding of, guideline projects. Drafts of the guideline have been reviewed by at least three AAN committees, at least one AANEM committee, a network of neurologists, Neurology peer reviewers,
and representatives from related fields. The AAN Guideline Author Conflict of Interest Policy can be viewed at www.aan.com. For complete information on this process, access the 2004 AAN process manual.e23
Appendix e-1. AAN GDDI members and mission
The mission of the GDDI is to develop, disseminate, and implement evidence-based systematic reviews and clinical practice guidelines related to the causation, diagnosis, treatment, and prognosis of neurologic disorders.
The GDDI is committed
to using the most rigorous methods available within its budget, in collaboration with other available AAN resources, to most efficiently accomplish this mission.
Cynthia Harden, MD (Chair); Steven R. Messé, MD (Co-Vice-Chair); Sonja
Potrebic, MD, PhD (Co-Vice-Chair); Eric J. Ashman, MD, FAAN; Richard L. Barbano, MD, PhD, FAAN; Brian Callaghan, MD; Jane Chan, MD, FAAN; Diane Donley, MD; Richard M. Dubinsky, MD, MPH, FAAN; Terry Fife, MD, FAAN; Jeffrey Fletcher, MD; Michael Haboubi,
MD; John J. Halperin, MD, FAAN; Yolanda Holler, MD; Andres M. Kanner, MD; Annette M. Langer-Gould, MD, PhD; Jason Lazarou, MD; Nicole Licking, DO; David Michelson, MD; Pushpa Narayanaswami, MBBS, DM, FAAN; Maryam Oskoui, MD; Richard Popwell, Jr.,
MD; Tamara Pringsheim, MD; Alejandro A. Rabinstein, MD, FAAN; Alexander Rae-Grant, MD; Anant Shenoy, MD; Kevin Sheth, MD, FAHA; Kelly Sullivan, PhD; Theresa A. Zesiewicz, MD, FAAN; Jonathan P. Hosey, MD, FAAN (Ex-Officio); Stephen Ashwal, MD, FAAN
(Ex-Officio); Deborah Hirtz, MD, FAAN; Jacqueline French, MD, FAAN (Guideline Process Historian)
Appendix e-2. AANEM Practice Issues Review Panel (PIRP) members
Yuen T. So, MD, PhD (Co-Chair); Williams S. David, MD, PhD (Co-Chair); Paul E. Barkhaus, MD; Earl J. Craig, MD; Prabhu D. Emmady, MD; Kenneth J. Gaines, MD; James F. Howard, MD; Atul T. Patel, MD; Bharathi Swaminathan, MD; Darrell T. Thomas, MD; Gil I.
Wolfe, MD
Appendix e-3. Complete search strategy
Original Search
Executed: October 2012
Databases: Medline, EMBASE, Cochrane, and Scopus databases




While the staff of HealthSearch makes every effort to ensure that the information gathered is accurate and up-to-date, HealthSearch disclaims any warranties regarding the accuracy or completeness of the information or its fitness for a particular purpose.
HealthSearch provides information from public sources both in electronic and print formats and does not guarantee its accuracy, completeness or reliability. The information provided is only for the use of the Client and no liability is accepted
by HealthSearch to third parties.
Scopus
Limited to 2011-2012
Query:
TITLE-ABS-KEY((muscular dystroph* AND (facioscapul* OR landouzy* OR dejerine* OR (erb W/2 dystroph*) OR (erb W/2 syndrom*) OR fsh OR fshd OR
fshmd))) AND SUBJAREA(mult OR agri OR bioc OR immu OR neur OR phar OR mult OR medi OR nurs OR vete OR dent OR heal OR mult OR arts OR busi OR deci OR econ OR psyc OR soci) AND PUBYEAR > 2010 AND PUBYEAR < 2013
Search results: 125
records
While the staff of HealthSearch makes every effort to ensure that the information gathered is accurate and up-to-date, HealthSearch disclaims any warranties regarding the accuracy or completeness of the information or its fitness for a
particular purpose. HealthSearch provides information from public sources both in electronic and print formats and does not guarantee its accuracy, completeness or reliability. The information provided is only for the use of the Client and no liability
is accepted by HealthSearch to third parties.
Updated Search
Executed: January 2014
Databases: Medline (via PubMed), and Cochrane
PubMed
Diagnosis/Broad[filter] AND ("muscular dystrophy, facioscapulohumeral"[MeSH Terms] OR ("muscular"[All Fields] AND "dystrophy"[All Fields] AND
"facioscapulohumeral"[All Fields]) OR "facioscapulohumeral muscular dystrophy"[All Fields] OR ("fascioscapulohumeral"[All Fields] AND "muscular"[All Fields] AND "dystrophy"[All Fields]) OR "fascioscapulohumeral muscular dystrophy"[All Fields])
Therapy/Broad[filter] AND ("muscular dystrophy, facioscapulohumeral"[MeSH Terms] OR ("muscular"[All Fields] AND "dystrophy"[All Fields] AND "facioscapulohumeral"[All Fields]) OR "facioscapulohumeral muscular dystrophy"[All Fields] OR ("fascioscapulohumeral"[All Fields]
AND "muscular"[All Fields] AND "dystrophy"[All Fields]) OR "fascioscapulohumeral muscular dystrophy"[All Fields])
Appendix e-4. AAN rules for classification of evidence for risk of bias
For questions related to screening (yield)
Class I
| - | Study of a cohort of patients at risk for the outcome from a defined geographic area (i.e., population based) |
| - | The outcome is objective |
| - | Also required: |
| a. Inclusion criteria defined | |
| b. At least 80% of patients undergo the screening of interest |
Class II
| - | A non-population-based, nonclinical cohort (e.g., mailing list, volunteer panel) or a general medical, neurology clinic/center without a specialized interest in the outcome. Study meets criteria a b (see Class I) |
| - | The outcome is objective |
Class III
| - | A referral cohort from a center with a potential specialized interest in the outcome |
Class IV
| - | Did not include persons at risk for the outcome |
| - | Did not statistically sample patients, or patients specifically selected for inclusion by outcome |
| - | Undefined or unaccepted screening procedure or outcome measure |
| - | No measure of frequency or statistical precision calculable |
For questions related to prognostic accuracy
Class I
| - | Cohort survey with prospective data collection |
| - | Includes a broad spectrum of persons at risk for developing the outcome |
| - | Outcome measurement is objective or determined without knowledge of risk factor status |
| - | Also required: |
| a. Inclusion criteria defined | |
| b. At least 80% of enrolled subjects have both the risk factor and outcome measured |
Class II
| - | Cohort study with retrospective data collection or case-control study. Study meets criteria a and b (see Class I) |
| - | Includes a broad spectrum of persons with and without the risk factor and the outcome |
| - | The presence of the risk factor and outcome are determined objectively or without knowledge of one another |
Class III
| - | Cohort or case-control study |
| - | Narrow spectrum of persons with or without the disease |
| - | The presence of the risk factor and outcome are determined objectively, without knowledge of the other or by different investigators |
Class IV
| - | Did not include persons at risk for the outcome |
| - | Did not include patients with and without the risk factor |
| - | Undefined or unaccepted measures of risk factor or outcomes |
| - | No measures of association or statistical precision presented or calculable |
For questions related to therapeutic intervention
Class I
| - | Randomized, controlled clinical trial (RCT) in a representative population |
| - | Masked or objective outcome assessment |
| - | Relevant baseline characteristics are presented and substantially equivalent between treatment groups, or there is appropriate statistical adjustment for differences |
| - | Also required: |
| a. Concealed allocation | |
| b. Primary outcome(s) clearly defined | |
| c. Exclusion/inclusion criteria clearly defined | |
| d. Adequate accounting for dropouts (with at least 80% of enrolled subjects completing the study) and crossovers with numbers sufficiently low to have minimal potential for bias | |
| e. For noninferiority or equivalence trials claiming to prove efficacy for one or both drugs, the following are also required*: |
| 1. | The authors explicitly state the clinically meaningful difference to be excluded by defining the threshold for equivalence or noninferiority | |
| 2. | The standard treatment used in the study is substantially similar to that used in previous studies establishing efficacy of the standard treatment (e.g., for a drug, the mode of administration, dose, and dosage adjustments are similar to those previously shown to be effective) | |
| 3. | The inclusion and exclusion criteria for patient selection and the outcomes of patients on the standard treatment are comparable to those of previous studies establishing efficacy of the standard treatment | |
| 4. | The interpretation of the study results is based on a per-protocol analysis that accounts for dropouts or crossovers |
Class II
| - | Cohort study meeting criteria a–e above or an RCT that lacks one or two criteria b–e |
| - | All relevant baseline characteristics are presented and substantially equivalent among treatment groups, or there is appropriate statistical adjustment for differences |
| - | Masked or objective outcome assessment |
Class III
| - | Controlled studies (including studies with external controls such as well-defined natural history controls) |
| - | A description of major confounding differences between treatment groups that could affect outcome** |
| - | Outcome assessment masked, objective, or performed by someone who is not a member of the treatment team |
Class IV
| - | Did not include patients with the disease |
| - | Did not include patients receiving different interventions |
| - | Undefined or unaccepted interventions or outcome measures |
| - | No measures of effectiveness or statistical precision presented or calculable |
*Numbers 1–3 in Class Ie are required for Class II in equivalence trials. If any one of the three is missing, the class is automatically downgraded to Class III
**Objective outcome measurement: an outcome measure that is unlikely to be affected
by an observer’s (patient, treating physician, investigator) expectation or bias (e.g., blood tests, administrative outcome data)
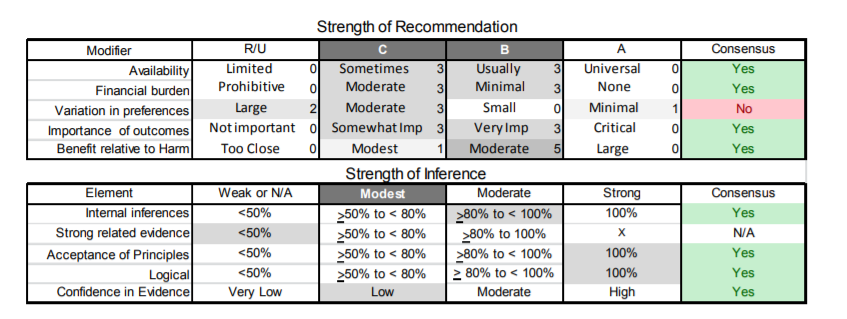
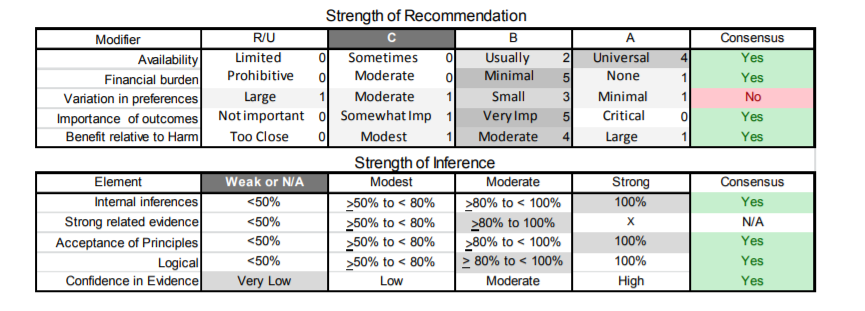
Appendix e-5. Steps and rules for formulating recommendations
Constructing the recommendation and its rationale
Rationale for recommendation summarized in the Clinical Context includes three categories of premises
- Evidence-based conclusions for the systematic review
- Stipulated axiomatic principles of care
- Strong evidence from related conditions not systematically reviewed
Actionable recommendations include the following mandatory elements
- The patient population that is the subject of the recommendation
- The person performing the action of the recommendation statement
- The specific action to be performed
- The expected outcome to be attained
Assigning a level of obligation
Modal modifiers used to indicate the final level of obligation (LOO)
- Level A: Must
- Level B: Should
- Level C: Might
- Level U: No recommendation supported
LOO assigned by eliciting panel members’ judgments regarding multiple domains, using a modified Delphi process. Goal is to attain consensus after a maximum of three rounds of voting. Consensus is defined by:
- > 80% agreement on dichotomous judgments
- >80% agreement, within one point for ordinal judgments
- If consensus obtained, LOO assigned at the median. If not obtained, LOO assigned at the 10th percentile
Three steps used to assign final LOO
1. Initial LOO determined by the cogency of the deductive inference supporting the recommendation on the basis of ratings within four domains. Initial LOO anchored to lowest LOO supported by any domain.
- Confidence in evidence. LOO anchored to confidence in evidence determined by modified form of the Grading of Recommendations Assessment, Development and Evaluation processe78
- Level A: High confidence
- Level B: Moderate confidence
- Level C: Low confidence
- Level U: Very low confidence
- Soundness of inference assuming all premises are true. LOO anchored to proportion of panel members convinced of soundness of the inference
- • Level A: 100%
- Level B: >80% to <100%
- Level C: >50% to <80%
- Level U or R: <50%
- Acceptance of axiomatic principles: LOO anchored to proportion of panel members who accept principles
- Level A: 100%
- Level B: >80% to <100%
- Level C: >50% to <80%
- Level U or R: <50%
- Belief that evidence cited from rerated conditions is strong: LOO anchored to proportion of panel members who believe the related evidence is strong
- Level B: >80% to 100% (recommendations dependent on inferences from non-systematically reviewed evidence cannot be anchored to a Level A LOO)
- Level C: >50% to <80%
- Level U or R: <50%
2. LOO is modified mandatorily on the basis of the judged magnitude of benefit relative to harm expected to be derived from complying with the recommendation
- Magnitude relative to harm rated on 4-point ordinal scale
- Large benefit relative to harm: benefit judged large, harm judged none
- Moderate benefit relative to harm: benefit judged large, harm judged minimal; or benefit judged moderate, harm judged none
- Small benefit relative to harm: benefit judged large, harm judged moderate; or benefit judged moderate, harm judged minimal; or benefit judged small, harm judged none
- Benefit to harm judged too close to call: Benefit and harm judged to be the same
- Regardless of cogency of the recommendation the LOO can be no higher than that supported by the rating of the magnitude of benefit relative to harm
- Level A: Large benefit relative to harm
- Level B: Moderate benefit relative to harm
- Level C: Small benefit relative to harm
- Level U: Too close to call
- LOO can be increased by one grade if LOO corresponding to benefit relative to harm greater than LOO corresponding to the cogency of the recommendation
3. LOO optionally downgraded on the basis of the following domains
- Importance of the outcome: critical, important, mildly important, not important
- Expected variation in patient preferences: none, minimal, moderate, large
- Financial burden relative to benefit expected: none, minimal, moderate, large
- Availability of intervention: universal, usually, sometimes, limited
Appendix e-6. Clinical contextual profiles
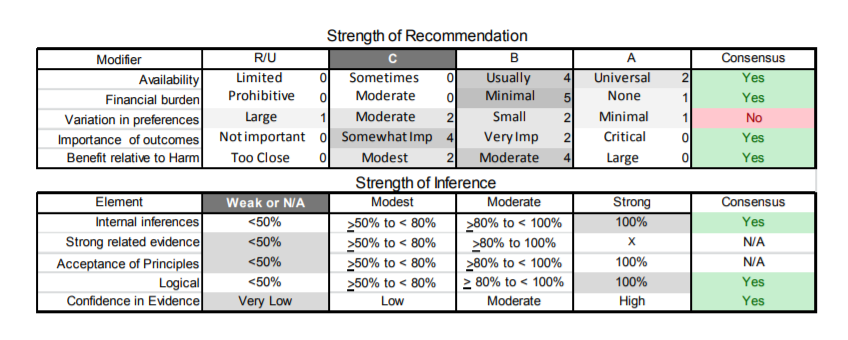
| A1. | Clinicians should obtain genetic confirmation of FSHD1 in patients with atypical presentations and no first-degree relatives with genetic confirmation of the disease (Level B). |
| B1. | Large D4Z4 deletion sizes (contracted D4Z4 allele of 10–20 kb) should alert the clinician that the patient is more likely to develop more significant disability and at an earlier age. Patients with large deletions are also more likely to develop symptomatic extramuscular manifestations (Level B). |
| C1. | Clinicians should obtain baseline pulmonary function tests on all patients with FSHD. Patients should be monitored regularly if they have abnormal baseline pulmonary function test results or any combination of severe proximal weakness, kyphoscoliosis, wheelchair dependence, or comorbid conditions that may affect ventilation (e.g., chronic obstructive pulmonary disease, cardiac disease) (Level B). |
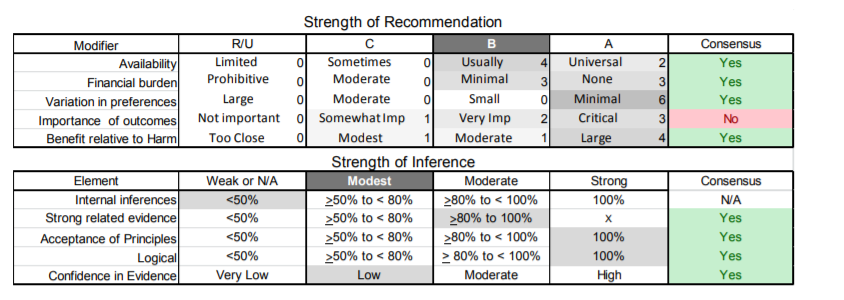
| C2. | In patients who have FSHD and compromised pulmonary function studies (e.g., FVC <60%) or symptoms of excessive daytime somnolence or nonrestorative sleep (e.g., frequent nocturnal arousals, morning headaches), clinicians should refer for pulmonary or sleep medicine consultation for consideration of nocturnal sleep monitoring or nocturnal noninvasive ventilation in order to improve QOL (Level B). |
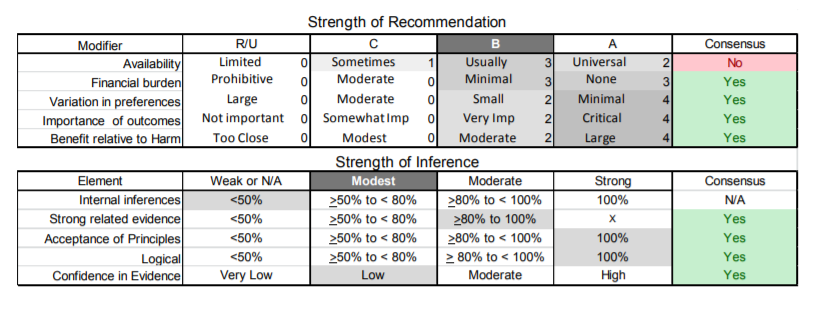
| C3. | Patients with FSHD who do not get regular pulmonary function testing should be tested prior to surgical procedures requiring general anesthesia, as such testing may uncover asymptomatic respiratory compromise (Level B). |
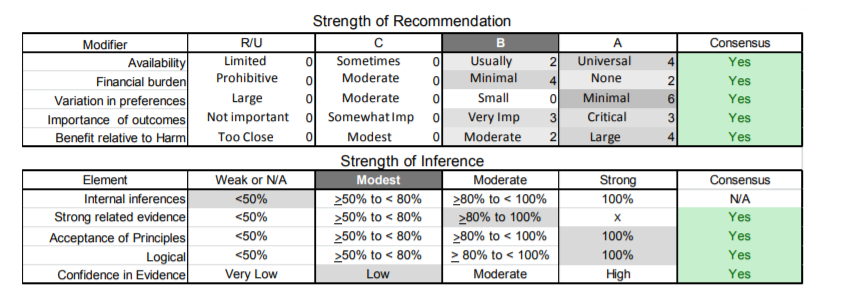
| C4. | Patients with FSHD should be referred for cardiac evaluation if they develop overt signs or symptoms of cardiac disease (e.g., shortness of breath, chest pain, palpitations). However, routine cardiac screening is not essential in the absence of cardiac signs or symptoms (Level C). |
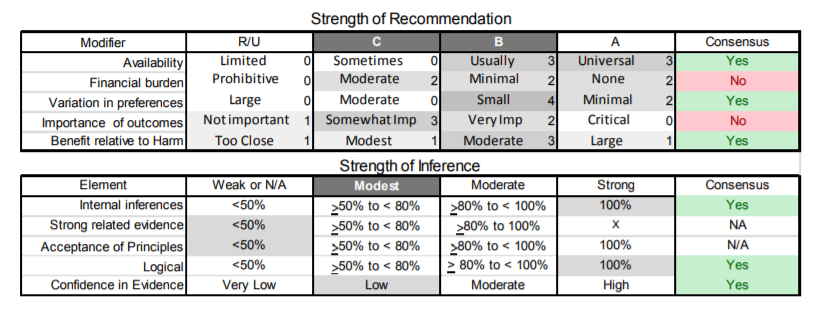
| C5. | Clinicians should refer patients with FSHD and large deletions (contracted D4Z4 allele of 10–20 kb) to an experienced ophthalmologist (e.g., retina specialist) for dilated indirect ophthalmoscopy (Level B). The presence and severity of retinal vascular disease at initial screening should be used to determine the frequency of subsequent monitoring (Level B). |
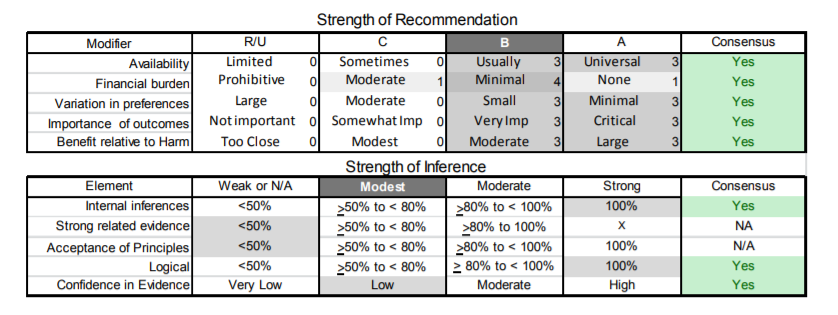
| C6. | Clinicians should screen all young children with FSHD at diagnosis and yearly thereafter until these children start school, as hearing loss may not be present at diagnosis and can be progressive (Level B). |
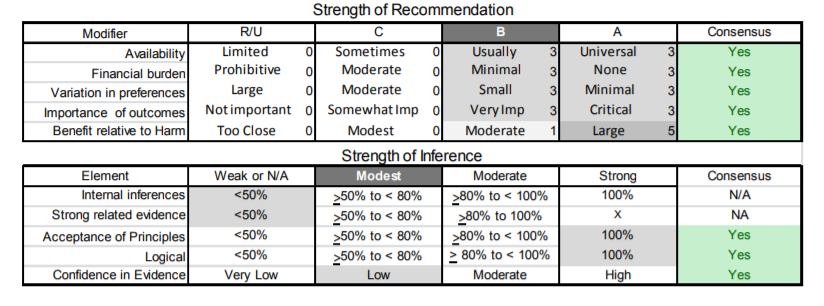
| C7. | Treating physicians should routinely inquire about pain in patients with FSHD. Referral for a physical therapy evaluation may prove helpful as an initial nonpharmacologic intervention. In patients with persistent pain and no contraindications, a trial of nonsteroidal anti-inflammatory medications is appropriate for acute pain and antidepressants or antiepileptics for chronic pain (Level B). |
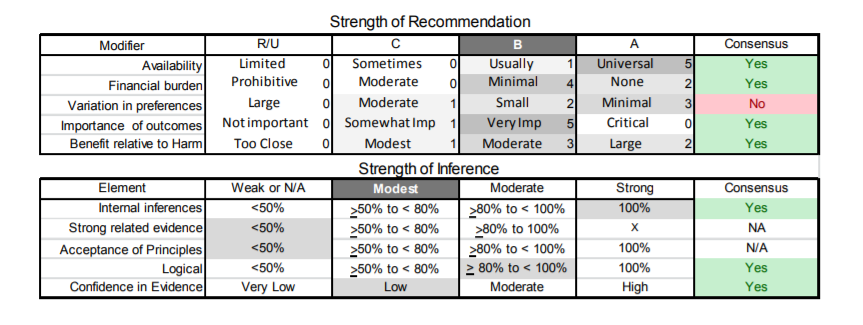
| D1. | In patients with FSHD, clinicians should not prescribe albuterol, corticosteroid, or diltiazem for improving strength (Level B). |
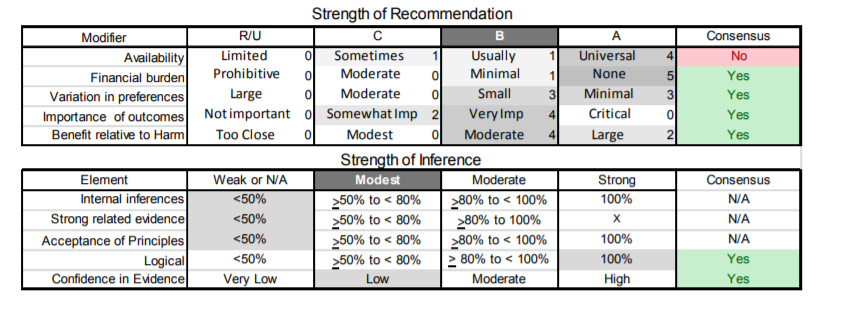
The author panel judged that even for therapies with only Class IV evidence, the known side effects (e.g., steroids) affected the risk–benefit tradeoff in such a way as to warrant a recommendation against use.
| D2. | Surgical scapular fixation might be offered cautiously to selected patients after careful consideration of the overall muscle impairment in the involved arm, assessment of potential gain in range of motion by manual fixation of the scapula, the patient’s rate of disease progression, and the potential adverse consequences of surgery and prolonged postsurgical bracing (Level C). |
| D3. | Clinicians might encourage patients with FSHD to engage in low-intensity aerobic exercise. An experienced physical therapist can help guide development of individualized exercise programs. Clinicians might also use the practical physical activities guidelines for individuals with disabilities, provided by the US Department of Health and Human Services, when counseling patients about aerobic exercise (Level C).e76 |
| D4. | In patients interested in strength training, clinicians may refer patients to physical therapists to establish a safe exercise program using appropriate low/medium weights/resistance that takes into consideration the patients’ physical limitations (Level C). |
References
| e1. | Padberg GW. Facioscapulohumeral disease. Leiden: University of Leiden; 1982. |
| e2. | Flanigan KM, Coffeen CM, Sexton L, Stauffer D, Brunner S, Leppert MF. Genetic characterization of a large, historically significant Utah kindred with facioscapulohumeral dystrophy. Neuromuscul Disord 2001;11:525–529. |
| e3. | Tawil R, Van Der Maarel SM. Facioscapulohumeral muscular dystrophy. Muscle Nerve 2006;34:1–15. |
| e4. | The FSH-DY Group. A prospective, quantitative study of the natural history of facioscapulohumeral muscular dystrophy (FSHD): implications for therapeutic trials. The FSH-DY Group. Neurology 1997;48:38–46. |
| e5. | Stübgen JP, Stipp A. Facioscapulohumeral muscular dystrophy: a prospective study of weakness and functional impairment. J Neurol 2010;257:1457–1464. |
| e6. | Wijmenga C, Brouwer OF, Padberg GW, Frants RR. Transmission of de-novo mutation associated with facioscapulohumeral muscular dystrophy. Lancet 1992;340:985–986. |
| e7. | van Deutekom JC, Wijmenga C, van Tienhoven EA, et al. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum Mol Genet 1993;2:2037–2042. |
| e8. | Lemmers RJ, de Kievit P, Sandkuijl L, et al. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat Genet 2002;32:235–236. |
| e9. | Gabriels J, Beckers MC, Ding H, et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 1999;236:25–32. |
| e10. | Lyle R, Wright TJ, Clark LN, Hewitt JE. The FSHD-associated repeat, D4Z4, is a member of a dispersed family of homeobox-containing repeats, subsets of which are clustered on the short arms of the acrocentric chromosomes. Genomics 1995;28:389–397. |
| e11. | Lemmers RJ, van der Vliet PJ, Klooster R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010;329:1650–1653. |
| e12. | Snider L, Geng LN, Lemmers RJ, et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet 2010;6:e1001181. |
| e13. | de Greef JC, Lemmers RJ, van Engelen BG, et al. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum Mutat 2009;30:1449– 1459. |
| e14. | de Greef JC, Lemmers RJ, Camaño P, et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology 2010;75:1548–1554. |
| e15. | Lemmers RJ, Tawil R, Petek LM, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet 2012;44:1370–1374. |
| e16. | Lemmers RJ, van der Vliet PJ, Klooster R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010;329:1650–1653. |
| e17. | Tawil R, van der Maarel SM, Tapscott SJ. Facioscapulohumeral dystrophy: the path to consensus on pathophysiology. Skelet Muscle 2014;4:12. |
| e18. | Tawil R, van der Maarel S, Padberg GW, van Engelen BG. 171st ENMC international workshop: Standards of care and management of facioscapulohumeral muscular dystrophy. Neuromuscul Disord 2010;20:471–475. |
| e19. | Attarian S, Salort-Campana E, Nguyen K, Behin A, Andoni Urtizberea J. Recommendations for the management of facioscapulohumeral muscular dystrophy in 2011. Rev Neurol (Paris) 2012;168:910–918. |
| e20. | Narayanaswami P, Weiss M, Selcen D, et al. Evidence-based guideline summary: Diagnosis and treatment of limb-girdle and distal dystrophies: report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology 2014;83:1453-1463. |
| e21. | Kang PB, Morrison L, Iannaccone ST, et al. Evidence-based guideline summary: Evaluation, diagnosis, and management of congenital muscular dystrophy. Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology. In press. |
| e22. | Padberg GW, Lunt PW, Koch M, Fardeau M. Diagnostic criteria for facioscapulohumeral muscular dystrophy. Neuromuscul Disord 1991;1:231–234. |
| e23. | American Academy of Neurology. Clinical Practice Guidelines Process Manual, 2004 ed. St. Paul, MN: The American Academy of Neurology; 2004. https://www.aan.com/Guidelines/Home/Development. Published 2004. Accessed February 12, 2012. |
| e24. | American Academy of Neurology. Clinical Practice Guidelines Process Manual, 2011 ed. St. Paul, MN: The American Academy of Neurology; 2011. https://www.aan.com/Guidelines/Home/Development. Published August 2011. Accessed February 12, 2012. |
| e25. | Hsu YD, Kao MC, Shyu WC, et al. Application of chromosome 4q35-qter marker (pFR-1) for DNA rearrangement of facioscapulohumeral muscular dystrophy patients in Taiwan. J Neurol Sci 1997;149:73–79. |
| e26. | Funakoshi M, Goto K, Arahata K. Epilepsy and mental retardation in a subset of early onset 4q35-facioscapulohumeral muscular dystrophy. Neurology 1998;50:1791–1794. |
| e27. | Zeng Y, Zhang C, Su Q. Gene diagnosis of facioscapulohumeral muscular dystrophy [in Chinese]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2001;18:213–215. |
| e28. | Goto K, Lee JH, Matsuda C, et al. DNA rearrangements in Japanese facioscapulohumeral muscular dystrophy patients: clinical correlations. Neuromuscul Disord 1995;5:201–208. |
| e29. | Scionti I, Greco F, Ricci G, et al. Large-scale population analysis challenges the current criteria for the molecular diagnosis of fascioscapulohumeral muscular dystrophy. Am J Hum Genet 2012;90:628–635. |
| e30. | Ricci E, Galluzzi G, Deidda G, et al. Progress in the molecular diagnosis of facioscapulohumeral muscular dystrophy and correlation between the number of KpnI repeats at the 4q35 locus and clinical phenotype. Ann Neurol 1999;45:751–757. |
| e31. | Lemmers RJ, Wohlgemuth M, van der Gaag KJ, et al. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am J Hum Genet 2007;81:884–894. |
| e32. | Spurlock G, Jim HP, Upadhyaya M. Confirmation that the specific SSLP microsatellite allele 4qA161 segregates with fascioscapulohumeral muscular dystrophy (FSHD) in a cohort of multiplex and simplex FSHD families. Muscle Nerve 2010;42:820–821. |
| e33. | Köhler J, Röhrig D, Bathke KD, Koch MC. Evaluation of the facioscapulohumeral muscular dystrophy (FSHD1) phenotype in correlation to the concurrence of 4q35 and 10q26 fragments. Clin Genet 1999;55:88–94. |
| e34. | Lunt PW, Jardine PE, Koch M, et al. Phenotypic-genotypic correlation will assist genetic counseling in 4q35-facioscapulohumeral muscular dystrophy. Muscle Nerve Suppl 1995;2:S103–S109. |
| e35. | Tawil R, Forrester J, Griggs RC, et al. Evidence for anticipation and association of deletion size with severity in facioscapulohumeral muscular dystrophy. The FSH-DY Group. Ann Neurol 1996;39:744–748. |
| e36. | Kilmer DD, Abresch RT, McCrory MA, et al. Profiles of neuromuscular diseases. Facioscapulohumeral muscular dystrophy. Am J Phys Med Rehabil 1995;74(5 Suppl):S131–S139. |
| e37. | Padua L, Aprile I, Frusciante R, et al. Quality of life and pain in patients with facioscapulohumeral muscular dystrophy. Muscle Nerve 2009;40:200–205. |
| e38. | Statland JM, Tawil R. Risk of functional impairment in facioscapulohumeral muscular dystrophy. Muscle Nerve 2014;49:520–527. |
| e39. | Sakellariou P, Kekou K, Fryssira H, et al. Mutation spectrum and phenotypic manifestation in FSHD Greek patients. Neuromuscul Disord 2012;22:339–349. |
| e40. | Wohlgemuth M, van der Kooi EL, van Kesteren RG, van der Maarel SM, Padberg GW. Ventilatory support in facioscapulohumeral muscular dystrophy. Neurology 2004;63:176–178. |
| e41. | Stübgen JP, Schultz C. Lung and respiratory muscle function in facioscapulohumeral muscular dystrophy. Muscle Nerve 2009;39:729–734. |
| e42. | de Visser M, de Voogt WG, la Rivière GV. The heart in Becker muscular dystrophy, facioscapulohumeral dystrophy, and Bethlem myopathy. Muscle Nerve 1992;15:591– 596. |
| e43. | Stevenson WG, Perloff JK, Weiss JN, Anderson TL. Facioscapulohumeral muscular dystrophy: evidence for selective, genetic electrophysiologic cardiac involvement. J Am Coll Cardiol 1990;15:292–299. |
| e44. | Galetta F, Franzoni F, Sposito R, et al. Subclinical cardiac involvement in patients with facioscapulohumeral muscular dystrophy. Neuromuscul Disord 2005;15:403–408. |
| e45. | Trevisan CP, Pastorello E, Armani M, et al. Facioscapulohumeral muscular dystrophy and occurrence of heart arrhythmia. Eur Neurol 2006;56:1–5. |
| e46. | Laforêt P, de Toma C, Eymard B, et al. Cardiac involvement in genetically confirmed facioscapulohumeral muscular dystrophy. Neurology 1998;51:1454–1456. |
| e47. | Nakagawa M, Matsuzaki T, Higuchi I, et al. Facioscapulohumeral muscular dystrophy: clinical diversity and genetic abnormalities in Japanese patients. Intern Med 1997;36:333–339. |
| e48. | Padberg GW, Brouwer OF, de Keizer RJ, et al. On the significance of retinal vascular disease and hearing loss in facioscapulohumeral muscular dystrophy. Muscle Nerve Suppl 1995;2:S73–S80. |
| e49. | Fitzsimons RB, Gurwin EB, Bird AC. Retinal vascular abnormalities in facioscapulohumeral muscular dystrophy. A general association with genetic and therapeutic implications. Brain 1987;110:631–648. |
| e50. | Statland JM, Sacconi S, Farmakidis C, Donlin-Smith CM, Chung M, Tawil R. Coats syndrome in facioscapulohumeral dystrophy type 1: frequency and D4Z4 contraction size. Neurology 2013;80:1247–1250. |
| e51. | Trevisan CP, Pastorello E, Ermani M, et al. Facioscapulohumeral muscular dystrophy: a multicenter study on hearing function. Audiol Neurootol 2008;13:1–6. |
| e52. | Rogers MT, Zhao F, Harper PS, Stephens D. Absence of hearing impairment in adult onset facioscapulohumeral muscular dystrophy. Neuromuscul Disord 2002;12:358–365. |
| e53. | Lutz KL, Holte L, Kliethermes SA, Stephan C, Mathews KD. Clinical and genetic features of hearing loss in facioscapulohumeral muscular dystrophy. Neurology 2013;81:1374– 1377. |
| e54. | Jensen MP, Hoffman AJ, Stoelb BL, Abresch RT, Carter GT, McDonald CM. Chronic pain in persons with myotonic dystrophy and facioscapulohumeral dystrophy. Arch Phys Med Rehabil 2008;89:320–328. |
| e55. | Guy-Coichard C, Nguyen DT, Delorme T, Boureau F. Pain in hereditary neuromuscular disorders and myasthenia gravis: a national survey of frequency, characteristics, and impact. J Pain Symptom Manage 2008;35:40–50. |
| e56. | van der Kooi EL, Vogels OJ, van Asseldonk RJ, et al. Strength training and albuterol in facioscapulohumeral muscular dystrophy. Neurology 2004;63:702–708. |
| e57. | Kissel JT, McDermott MP, Mendell JR, et al; FSH-DY Group. Randomized, double-blind, placebo-controlled trial of albuterol in facioscapulohumeral dystrophy. Neurology 2001;57:1434–1440. |
| e58. | Wagner KR, Fleckenstein JL, Amato AA, et al. A phase I/II trial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol 2008;63:561–571. |
| e59. | van der Kooi EL, Kalkman JS, Lindeman E, et al. Effects of training and albuterol on pain and fatigue in facioscapulohumeral muscular dystrophy. J Neurol 2007;254:931–940. |
| e60. | Tawil R, McDermott MP, Pandya S, et al. A pilot trial of prednisone in facioscapulohumeral muscular dystrophy. FSH-DY Group. Neurology 1997;48:46–49. |
| e61. | Elsheikh BH, Bollman E, Peruggia M, King W, Galloway G, Kissel JT. Pilot trial of diltiazem in facioscapulohumeral muscular dystrophy. Neurology 2007;68:1428–1429. |
| e62. | Copeland SA, Levy O, Warner GC, Dodenhoff RM. The shoulder in patients with muscular dystrophy. Clin Orthop Relat Res 1999;(368):80–91. |
| e63. | Rhee YG, Ha JH. Long-term results of scapulothoracic arthrodesis of facioscapulohumeral muscular dystrophy. J Shoulder Elbow Surg 2006;15:445–450. |
| e64. | Diab M, Darras BT, Shapiro F. Scapulothoracic fusion for facioscapulohumeral muscular dystrophy. J Bone Joint Surg Am 2005;87:2267–2275. |
| e65. | Krishnan SG, Hawkins RJ, Michelotti JD, Litchfield R, Willis RB, Kim YK. Scapulothoracic arthrodesis: indications, technique, and results. Clin Orthop Relat Res 2005;(435):126–133. |
| e66. | Jakab E, Gledhill RB. Simplified technique for scapulocostal fusion in facioscapulohumeral dystrophy. J Pediatr Orthop 1993;13:749–751. |
| e67. | Andrews CT, Taylor TC, Patterson VH. Scapulothoracic arthrodesis for patients with facioscapulohumeral muscular dystrophy. Neuromuscul Disord 1998;8:580–584. |
| e68. | Giannini S, Faldini C, Pagkrati S, et al. Fixation of winged scapula in facioscapulohumeral muscular dystrophy. Clin Med Res 2007;5:155–162. |
| e69. | Giannini S, Ceccarelli F, Faldini C, Pagkrati S, Merlini L. Scapulopexy of winged scapula secondary to facioscapulohumeral muscular dystrophy. Clin Orthop Relat Res 2006;449:288–294. |
| e70. | Demirhan M, Uysal O, Atalar AC, Kilicoglu O, Serdaroglu P. Scapulothoracic arthrodesis in facioscapulohumeral dystrophy with multifilament cable. Clin Orthop Relat Res 2009;467:2090–2097. |
| e71. | Bunch WH, Siegel IM. Scapulothoracic arthrodesis in facioscapulohumeral muscular dystrophy. Review of seventeen procedures with three to twenty-one-year follow-up. J Bone Joint Surg Am 1993;75:372-376. |
| e72. | Berne D, Laude F, Laporte C, Fardeau M, Sailant G. Scapulothoracic arthrodesis in facioscapulohumeral muscular dystrophy. Clin Orthop Relat Res 2003;409:106-113. |
| e73. | Olsen DB, Ørngreen MC, Vissing J. Aerobic training improves exercise performance in facioscapulohumeral muscular dystrophy. Neurology 2005;64:1064–1066. |
| e74. | Ambrosino N, Carpenè N, Gherardi M. Chronic respiratory care for neuromuscular diseases in adults. Eur Respir J 2009;34:444–451. |
| e75. | Voet NB, van der Kooi EL, Riphagen II, Lindeman E, van Engelen BG, Geurts AC. Strength training and aerobic exercise training for muscle disease. Cochrane Database Syst Rev 2013;7:CD003907. |
| e76. | US Department of Health and Human Services Office of Disease Prevention and Health Promotion. Additional Considerations for Some Adults. In: Physical Activity Guidelines for Americans;chap 7. http://www.health.gov/paguidelines/guidelines/chapter7.aspx.Published September 2008. Accessed October 1, 2012. |
| e77. | von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP; for the STROBE Initiative. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. J Clin Epidemiol. 2008;61:344-349. |
| e78. | Guyatt GH, Oxman AD, Schünemann HJ, Tugwell P, Knottnerus A. GRADE guidelines: A new series of articles in the Journal of Clinical Epidemiology. J Clin Epidemiol 2011;64:380–382. |
Document History
This guideline was endorsed by the FSH Society on December 18, 2014.
Creation of New Guidelines, Consensus Statements, or Position Papers
AANEM members are encouraged to submit ideas for papers that can improve the understanding of the field. The AANEM will review nominated topics on the basis of the following criteria:- Members’ needs
- Prevalence of condition
- Health impact of condition for the individual and others
- Socioeconomic impact
- Extent of practice variation
- Quality of available evidence
- External constraints on practice
- Urgency for evaluation of new practice technology