Evidence-Based Guideline: Diagnosis and Treatment of Limb-Girdle and Distal Dystrophies
Report of the Guideline Development Subcommittee of the American Academy of
Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine
Pushpa Narayanaswami, MBBS, DM, FAAN1; Michael Weiss, MD, FAAN2; Duygu Selcen,
MD3; William David, MD, PhD4; Elizabeth Raynor, MD1; Gregory Carter, MD5; Matthew Wicklund, MD, FAAN6; Richard J. Barohn, MD, FAAN7; Erik Ensrud, MD8,10; Robert C.
Griggs, MD, FAAN9; Gary Gronseth, MD, FAAN7; Anthony A. Amato, MD, FAAN10
- Department of Neurology, Beth Israel Deaconess Medical Center/Harvard Medical School, Boston, MA
- Department of Neurology, University of Washington Medical Center, Seattle, WA
- Department of Neurology, Mayo Clinic, Rochester, MN
- Department of Neurology, Massachusetts General Hospital, Boston, MA/Harvard Medical School, Boston, MA
- St Luke's Rehabilitation Institute, Spokane, WA
- Department of Neurology, Penn State Hershey Medical Center, Hershey, PA
- Department of Neurology, University of Kansas Medical Center, Kansas City, KS
- Neuromuscular Center, Boston VA Medical Center, Boston, MA
- Department of Neurology, University of Rochester Medical Center, Rochester, NY
- Department of Neurology, Brigham and Women’s Hospital, Boston, MA/Harvard Medical School, Boston, MA
ABSTRACT
Objective: To review the current evidence and make practice recommendations regarding the diagnosis and treatment of limb-girdle muscular dystrophies (LGMDs).
Methods: Systematic review and practice recommendation development using the American Academy of Neurology guideline development process.
Results: Most LGMDs are rare, with estimated prevalences ranging from 0.07 per 100,000 to 0.43 per 100,000. The frequency of some muscular dystrophies varies based on the ethnic background of the population studied. Some LGMD subtypes have distinguishing features, including pattern of muscle involvement, cardiac abnormalities, extramuscular involvement and muscle biopsy findings. The few published therapeutic trials were not designed to establish clinical efficacy of any treatment.
Principal Recommendations: For patients with suspected muscular dystrophy, clinicians should use a clinical approach to guide genetic diagnosis based on clinical phenotype, inheritance pattern, and associated manifestations (Level B). Clinicians should refer newly diagnosed patients with an LGMD subtype and high risk of cardiac complications for cardiology evaluation even if they are asymptomatic from a cardiac standpoint (Level B). In LGMD patients with a known high risk of respiratory failure, clinicians should obtain periodic pulmonary function testing (Level B). Clinicians should refer patients with muscular dystrophy to a clinic that has access to multiple specialties designed specifically to care for patients with neuromuscular disorders (Level B). Clinicians should not offer patients with LGMD gene therapy, myoblast transplantation, neutralizing antibody to myostatin, or growth hormone outside of a research study designed to determine efficacy and safety of the treatment (Level R).
INTRODUCTION
Limb-girdle muscular dystrophies (LGMDs) are a group of myopathies characterized by predominantly proximal muscle weakness (pelvic and shoulder girdles).e1 Initially described as a clinical phenotype, they are now recognized as a heterogeneous group of myopathies that vary in severity and may affect persons at all ages from childhood through adulthood. In 1995, the LGMDs were classified into 2 main groups depending on the inheritance pattern: LGMD1, autosomal dominant, and LGMD2, autosomal recessive. Overlaid on this numeric division is a letter designating the order of discovery for each chromosomal locus (e.g., LGMD1A implying autosomal dominant LGMD type A; LGMD2D implying autosomal recessive LGMD type D).e2,e3 With advances in molecular genetics that identify new genetic defects associated with the LGMD phenotype, this list of disorders continues to grow. Unfortunately, the literature is conflicting as to the appropriate terminology for different disorders. For example, prior to genetic discovery, and even after, various reports refer to some of these disorders as congenital myopathies, myofibrillar myopathies, hereditary inclusion body myopathies (hIBMs), distal myopathies/dystrophies, or LGMD.e4 Table e-1 delineates the most recent classification of what is considered “muscular dystrophies” in adults that were included in this review.
The LGMDs are rare disorders with a combined minimum prevalence of 2.27/100,000.e5 Given the wide variation in phenotypic expression of the LGMDs, establishing a clinical diagnosis is a challenge. Importantly, some of these disorders are associated with potentially serious cardiac and respiratory complications. In the evaluation of a patient with LGMD, the ideal approach is to utilize the person’s clinical presentation and narrow down the possible genotype to a few disorders. This will help both to predict the long-term prognosis and to plan further evaluation, such as muscle biopsy or blood tests to confirm the genetic defect and tests of cardiorespiratory function. With increasingly accurate molecular diagnosis, knowledge regarding the genotype/phenotype correlations, although far from complete, is slowly advancing. Although there is some literature discussing the clinical approach to LGMDs,e6 no systematic reviews of the literature or practice guidelines are available for clinicians who evaluate these disorders. This evidence-based guideline reviews the current evidence regarding the diagnosis and treatment of LGMDs.
We have classified the LGMDs by their molecular diagnosis and also discuss non–limb-girdle adult-onset myopathies that are genotypically identical to the LGMDs, such as Miyoshi distal myopathy, which is allelic to LGMD2B. In addition, other hereditary myopathies that overlap and may indeed be considered forms of LGMD (e.g., hIBMs, myofibrillar myopathies, Emery-Dreifuss muscular dystrophy [EDMD], Becker muscular dystrophy [BMD], manifesting carriers of dystrophin mutations) are included. We also review the distal myopathies. Hence, this review encompasses 3 major phenotypic dystrophies: limb-girdle weakness, humeroperoneal weakness as in EDMD, and distal weakness as in the distal myopathies. We use the terms LGMD and muscular dystrophy interchangeably to refer to the disorders reviewed in this guideline. Duchenne dystrophy, congenital muscular dystrophy, myotonic dystrophy, and facioscapulohumeral dystrophy are not included in this guideline, as they will be discussed in forthcoming guidelines. This guideline seeks to answer the following clinical questions:
- In a population of patients with suspected muscular dystrophy, what proportion of patients has a genetic defect confirming LGMD/distal myopathy/distal muscular dystrophy/BMD?
- In patients with muscular dystrophy, what is the association between specific features and subtypes of these disorders, in particular ethnicity; age at onset; scapular winging; weakness, atrophy, hypertrophy, or MRI changes in the facial muscles, calf, gastrocnemius, quadriceps, hip adductors, hip abductors, and tibialis anterior; cardiac dysfunction (arrhythmias, congestive heart failure, reduced/abnormal ejection fraction [EF], dilated cardiomyopathy, hypertrophic cardiomyopathy); respiratory dysfunction (abnormal/reduced forced vital capacity [FVC]); dysphagia; dysarthria; hoarse voice; contractures; and cognitive dysfunction? In patients with LGMD, what is the association between the degree of creatine kinase (CK) elevations and specific subtypes of these disorders, in particular CK normal, <10-fold elevation, and >10-fold elevation?
- In patients with LGMD or distal muscular dystrophy, what is the association between specific muscle biopsy features and subtypes of these disorders, in particular rimmed vacuoles, inflammation, and inclusions?
- How often do patients with muscular dystrophy and its specific subtypes have significant respiratory abnormalities (FVC < 50% predicted), cardiac abnormalities (EF < 50%, evidence of hypertrophic cardiomyopathy or generalized wall motion abnormality, arrhythmias, conduction defects), or bone loss (osteoporosis or bone mineral density 2.5 SD below peak bone mass, osteopenia or bone mass of 1.0–2.5 SD below peak bone mass)?
- Are there effective therapies (medications, gene therapy, exercise, complementary and alternative therapies, orthopedic interventions, surgery) for muscular dystrophies that improve muscle strength, slow the rate of strength decline, preserve ambulation and overall function, delay time to tracheostomy ventilation, maintain healthy EF, slow cardiac mortality, preserve quality of life and activities of daily living, and delay overall mortality?
DESCRIPTION OF THE ANALYTIC PROCESS
In July 2010, the American Academy of Neurology (AAN) Guideline Development Subcommittee and the American Association of Neuromuscular & Electrodiagnostic Medicine Practice Issues Review Panel (appendices e-1-e-3) formed a panel of neurologists, other physicians with relevant expertise, methodologists, and patient advocates. The MEDLINE, EMBASE, and Cochrane databases were searched from 1987 onward for relevant peer-reviewed articles in humans and in English only (appendix e-4 provides the full search strategy and terms). Through an initial search conducted in 2011 and an updated search conducted in 2013, a total of 3,246 abstracts were identified. Of those, 1,335 articles were selected for full-text review. Two panel members, working independently of each other, reviewed each of the 1,335 articles and selected 699 for final review and classification. Each final article was reviewed by 2 panel members who rated it according to the AAN 2011 criteria for classification of articles (appendix e-5), using the scheme appropriate to the clinical question. The AAN population screening evidence scheme was used for questions 1–4, and the therapeutic scheme for question 5. Where differences in article ratings occurred, a third panel member determined the ultimate rating. Recommendations were developed by a modified Delphi process, and ratings of the recommendations (appendix e-6) were linked to the strength of the evidence as per the 2011 guideline development process.e7 The recommendations are made by first assigning a confidence level to the evidence relative to each outcome that is deemed important. The confidence level depends on the class of studies available. The level of confidence is high if there are 2 Class I studies and very low if there are less than 2 Class III studies. Second, transparently discussed deductive principles and inferences are used to refine the level of recommendation. For instance, a Level B recommendation may be made if deductive inferences are convincing (>80% of the panel accepts them) as long as the confidence in the evidence is at least low (2 Class III studies).
Articles with descriptions of at least 3 patients were considered for inclusion. In instances of the initial description of a disorder, rare disorders, or rare manifestations of a disorder, we included studies with fewer than 3 patients. Studies were excluded if they reported group outcomes for more than one disorder and individual disorders could not be identified within the group. Genetic testing was necessary for confirmation of all diagnoses except BMD or manifest carriers of Duchenne muscular dystrophy (DMD), for which we accepted muscle biopsy immunohistochemistry/Western blot confirmation. Most often, the initial article describing the disorder did not have the gene defect identified, and therefore the article was not classified. However, these cross-referenced articles were reviewed in conjunction with the subsequent articles delineating the gene defect to obtain details of the clinical phenotype. For all questions, we classified the evidence by specific diseases: LGMD types 1A–E and 2A–P (autosomal dominant and recessive, respectively, where the gene/protein defects are known), distal myopathies, myofibrillar myopathies, EDMD, and hIBM. Because some LGMD gene defects may cause different phenotypes, the different disorders that are associated with the same gene defect are discussed together. It is also known that some protein defects can cause more severe phenotypes presenting early in childhood with congenital muscular dystrophy with or without brain involvement. We briefly state this when applicable but do not describe these phenotypes, as they will be addressed in forthcoming guidelines. We recognize that this classification is inherently artificial, because the phenotypes may merge with time. See page 174 of this document for an index of the diseases reviewed in this guideline and the pages on which they are discussed. Table e-1 and appendices e-1 through e-6 are available herein; figures e-1 and e-2 and table e-2 are available on the Neurology® Web site at Neurology.org.
ANALYSIS OF EVIDENCE
Clinical Question 1: In a population of patients with suspected muscular dystrophy, what proportion of patients has a genetic defect confirming LGMD/distal myopathy/distal muscular dystrophy/BMD?
No articles were available for disorders due to genetic defects in DNAJB6, TRIM32, FHL1, MYH7, filamin C, VCP, matrin-3, selenoprotein, cavin, nebulin, nesprin, KLHL9, and Welander distal myopathies.
LGMD1A (myotilin).
This is discussed below in the section on myofibrillar myopathies.
LGMD1B (lamin A/C, also causes autosomal dominant [AD]-EDMD).
One Class Ie5 and 9 Class III studiese8-e16 were reviewed. In a Class I population study of 1,105 patients with various genetic disorders of muscle, the frequency of LGMD1B/AD-EDMD was 8.8% (95% confidence interval [CI] 2.1–15.6), translating to a population prevalence of 0.2/100,000 (95% CI 0–0.4).e5
In the 9 Class III studies, the frequency of laminopathy among patients with LGMD ranged from 0.9-4%. However, when looking at patients with idiopathic cardiomyopathy, mutations in lamin A/C were found in 8%–39% of cases.
LGMD1C (caveolin-3).
Three Class III studiese11,e13,e17 were reviewed. Caveolin-3 mutations were identified in 1.3%-2.6% of patients in these series.
LGMD1E (desmin).
This is discussed below in the section on myofibrillar myopathies.
LGMD2A (calpain-3).
There were 2 Class I studiese5,e18 and 19 Class III studies.e11,e13,e17,e19-e34 Calpainopathies have been reported in patients of many ethnic backgrounds and from 6 continents. In a Class I study, the overall prevalence of calpainopathy among various genetic disorders of muscle was 0.6/100,000 (95% CI 0.3–0.9) and the prevalence among all LGMD cases was 18/68 or 26.5% (95% CI 16–37).e5 In the other Class I study of 84 Italian patients with an unknown muscular dystrophy, 39 patients (46.4%) had calpainopathy and the prevalance was calculated to be 9.47 per million.e18
In the 19 Class III studies, calpain-3 mutations accounted for 6%–57% of the LGMD, with the majority of series reporting 18.5%-35% of LGMD being calpainopathies. LGMD2A appeared to be the most common LGMD subtype in many published series in the Netherlands, England, Italy, Bulgaria, Spain, France, Turkey, Brazil, and Japan, and constituted 28.4% of known LGMD cases in northern Italy,e17 26.5% in northern England,e5 and 21% in the Netherlands.e34
LGMD2B (dysferlin).
One Class I studye5 and 11 Class III studiese10,e11,e13,e17,e19,e20,e23,e24,e28,e35,e36 were reviewed. Two studies describe the same cohort,e20,e24 with additional patients studied over time; therefore, the studies are reviewed together. A Class I studye5 found the prevalence of dysferlin mutations to be 0.13/100,000. The total group was composed of 1,105 patients with various hereditary muscle diseases, of which LGMDs overall constituted 6.15% (68 cases). Dysferlinopathies comprised 4/68 (5.9%, 95% CI 0.3–11.5) LGMD cases. The 11 Class III studies reported a frequency of dysferlinopathy ranging from 0.6%–33% of the LGMDs.
LGMD2C (γ-sarcoglycan).
Two Class Ie5,e37 and 16 Class IIIe10,e11,e13,e17,e19,e23,e24,e38-e46 studies were reviewed. A Class I studye5 found the overall prevalence of γ-sarcoglycanopathy to be 1.3 per 1 million (0.13/100,000; 95% CI 0–0.3), forming 5.9% (95% CI 0.3–11.5) of the 1,105 patients with genetic muscle diseases. Another Class I study evaluated the genetic–epidemiologic aspects of primary sarcoglycanopathies in a geographic area in northeast Italy between 1982 and 1996.e37 Muscle biopsies consistent with dystrophy and normal dystrophin were included in the analysis. Thirteen of 204 patients had a gene defect in one of the sarcoglycans. Four of the 13 were found to have γ-sarcoglycan gene defects (2 unrelated, 2 siblings) (4/204, 2%). The LGMD2C prevalence was 1.72 per 1 million.
The frequency of γ-sarcoglycanopathy in the 16 Class III studies ranged from 1.3%–13.2%. In those cases selected for abnormal expression of the sarcoglycans on immunohistochemistry, γsarcoglycan mutations were felt to be responsible in 7%–21%.
LGMD2D (α-sarcoglycan).
Two Class Ie5,e37 and 14 Class IIIe10,e13,e17,e23,e24,e38-e40,e43-e48 studies were reviewed. One Class I studye5 found the prevalence of α-sarcoglycanopathy to be 0.07 (95% CI 0–0.2) per 100,000. Another Class I study from Italy reported 7/204 (3.4%) patients as having α-sarcoglycan mutations. The prevalence of LGMD2C was 3.02 per million.e37 The 14 Class III studies reported α-sarcoglycan mutations to be responsible for 3.3%–15% of LGMDs. Of those with reduced expression of sarcoglycans on immunohistochemistry staining, 34%–40% were deemed to be LGMD2D.
LGMD2E (ß-sarcoglycan).
Two Class Ie5,e37 and 13 Class IIIe10,e11,e13,e17,e20,e23,e24,e38-e40,e43,e45,e46 studies were reviewed. One Class I studye5 found the prevalence of β-sarcoglycanopathies to be 0.07/100,000. β-Sarcoglycanopathies comprised 2.9% of the total group of genetic muscle diseases (1,105), of which LGMD formed 6.15% (68 cases). Another Class I study evaluated the genetic–epidemiologic aspects of sarcoglycanopathies in a geographic area in northeast Italy.e37 Two unrelated patients (2/204, 1%) were found to have β-sarcoglycan mutations; the prevalence of LGMD2E was 0.86/1000000.e37 The 13 Class III studies reported β-sarcoglycan mutations to be responsible for 0%–23% of LGMDs, with most reporting about 4%. Of those with reduced expression of sarcoglycans on immunohistochemistry staining, 15%–43% were deemed to be LGMD2E.
LGMD2F (δ-sarcoglycan).
Two Class Ie5,e37 and 12 Class IIIe10,e13,e17,e23,e24,e40,e43-e46,e49,e50 studies were reviewed. Neither Class I study reported δ-sarcoglycan mutations. The 12 Class III studies reported δ-sarcoglycan mutations to be responsible in 0%–14% of LGMDs, and approximately 8% of those cases had reduced sarcoglycan expression on immunohistochemistry.
LGMD2G (telethonin).
Two Class III studies were reviewed.e23,e51 In one Class III study of 63 unrelated patients with myofibrillar myopathy diagnosed by demonstration of myofibrillar degradation products and ectopic expression of multiple proteins on muscle biopsy, no mutations in the gene for telethonin were found.e51 In another Class III study of 140 patients with LGMD from 40 families, telethonin mutations were shown in 6 patients (4.2%) in one family (2.5%).e23
LGMD2I (FKRP).
One Class I study,e5 one Class II study,e52 and 12 Class III studiese10,e11,e13,e17,e28,e32,e46,e53-e57 were reviewed. The Class I studye5 found the prevalence of autosomal recessive FKRP mutations to be 0.43/100,000 (95% CI 0.2–0.7). Mutations involving FKRP were demonstrated in 19.1% (95% CI 9.8–28.5) of all genetic muscle diseases (1,105), 68 (6.15%) of which were LGMD. LGMD2I formed 19% of the LGMD group. In the Class II study, 2.0% (2/102) of consecutive unrelated German patients with persistent hyperCKemia were asymptomatic or minimally symptomatic (myalgia or fatigue), and 5.1% (5/98) of consecutive unrelated patients with LGMD2 had mutations in the FKRP gene.e52 The 12 Class III studies found mutations in the FKRP gene in 4%–30% of LGMD.
LGMD2J (titin).
One Class III study of 25 families and 25 sporadic cases of mainly distal myopathies revealed mutations in the titin gene in 4/25 (16%) families but in none of the sporadic cases.e58
LGMD2K (POMT1).
One Class III study of 92 patients with evidence of dystroglycanopathy based on muscle biopsy but negative genetic testing for FKRP mutations demonstrated that 8 patients (8.7%) had mutations in the POMT1 gene.e59 These included the following phenotypes and distributions: Walker-Warburg syndrome (WWS) (1/8), muscle-eye-brain disease/Fukuyama congenital muscular dystrophy (MEB/FCMD) (1/8), congenital muscular dystrophy with intellectual disability (mental retardation) (CMD-MR) (3/8), and LGMD with intellectual disability (mental retardation) (LGMD-MR) (3/8).
LGMD2L (anoctamin-5).
Two Class III studies were identified.e60,e61 In one Class III study of 64 British and German patients from 59 families with either a limb-girdle or Miyoshi myopathy phenotype without dysferlin mutations, 20 patients (31.3%) from 15 (25.4%) families had a mutation in the anoctamin-5 gene.e60 In another Class III study of 101 Finnish patients with undetermined LGMD, calf distal myopathy, or CK elevations more than 2,000 IU/L, 25 patients (24.8%) were identified with anoctamin-5 gene mutations.e61
LGMD2M (fukutin).
One Class III study of 92 patients with evidence of dystroglycanopathy based on muscle biopsy but negative genetic testing for FKRP mutations demonstrated that 6 patients (6.5%) had mutations in the fukutin gene.e59 These included the following phenotypes and distributions: WWS (1/6), MEB/FCMD (1/6), CMD without intellectual disability (mental retardation) (CMD-noMR) (1/6), and LGMD without intellectual disability (mental retardation) (LGMD-noMR) (3/6).
LGMD2N (POMT2).
One Class III study of 92 patients with evidence of dystroglycanopathy based on muscle biopsy but negative genetic testing for FKRP mutations demonstrated that 9 patients (9.7%) had mutations in the POMT2 gene.e59 These included the following phenotypes and distributions: MEB/FCMD (6/9), CMD with cerebellar ataxia (2/9), and LGMD-MR (1/9).
LGMD2O (POMGNT1).
One Class III study of 92 patients with evidence of dystroglycanopathy based on muscle biopsy but negative genetic testing for FKRP mutations demonstrated that 7 patients (7.6%) had mutations in the POMGNT1 gene.e59 Six of the 7 patients had MEB/FCMD and 1 had LGMD-noMR.
BMD.
Five Class Ie5,e62-e65 and 5 Class IIIe10,e66-e69 studies were reviewed. One Class I study identified 79 patients with BMD residing in the Northern Health Region of England by searching the clinical and muscle biopsy records.e62 The minimum prevalence was estimated to be 2.38/100,000. A Class I epidemiologic study in the territory of Northwest Tuscany, central Italy, estimated the incidence of BMD to be 2.42 x 10-5 male live births.e63 Thirty-one percent of patients with LGMD (32/103) from 29 families were found to be affected by BMD. Another
Class I study examined the prevalence of BMD in a geographically isolated area of Okinawa, Japan.e64 The prevalence was estimated to be 1.82 x 10-5 in the male population. The incidence of BMD in the period from 1957–1985 was 3.21 x 10-5 live-born males. However, this study may underestimate the prevalence and incidence because patients with BMD were diagnosed only on the basis of immunohistochemical analysis of muscle biopsies; Western blots were not performed. In another Class I study, 109 of 1,105 (9.9%, 95% CI 8.1–11.6) patients with inherited muscle diseases carried the diagnosis of BMD, with an estimated prevalence of 7.29/100,000 males (95% CI 5.9–8.7).e5 The last Class I study analyzed 3,048 muscle biopsies processed by the National Institute of Neuroscience in Tokyo and identified 41 patients as having LGMD.e65 Among those, 5 patients (12%) had BMD. Population prevalence was not provided. The 5 Class III studies reported the frequency of BMD to be 1.6%–55.6% of patients presenting with limb-girdle weakness.
Duchenne/Becker manifesting carriers.
Four studies, 2 Class Ie5,e65 and 2 Class III,e67,e70 were reviewed. One Class I studye5 found the prevalence of Duchenne/Becker manifest carriers to be 13/1,105 (1.2%) (95% CI 0.5–1.8), corresponding to a population prevalence of 0.43/100,000 (95% CI 0.2–0.7). In one Class I study of 3,048 Japanese patients with a diagnosis of LGMD based on clinical and histopathologic criteria, only 2 women had evidence of dystrophinopathy on immunohistochemistry.e65 In one of the Class III studies, in which 201 biopsies were reanalyzed using dystrophin immunoblot, 1/4 females with unclassified congenital myopathies (25%), 1/20 females with unclassified myopathies (5%), and 5/9 females (56%) with hyperCKemia were diagnosed as being manifest dystrophinopathy carriers.e67 The other Class III study retrospectively looked at 169 Israeli families with members affected by progressive muscular dystrophy. Molecular analysis was performed on 106 DMD and 5 BMD families, with 81 available probands. The investigators were able to exclude a diagnosis of DMD/BMD on the basis of clinical symptoms and signs (49 families), or normal dystrophin on biopsy and/or the absence of linkage to chromosome X by analysis of restriction fragment length polymorphism– derived haplotypes (11 families).e70
Emerin.
Two Class III studies were reviewed.e10,e13 In one study of 550 patients with the clinical diagnosis of childhood or adult LGMD, distoproximal myopathy, or hyperCKemia, emerin mutations were seen in 2/550 (0.4%). There were 346 patients with LGMD, for a frequency of 0.6% of all LGMD.e13 Another study found 2 of 370 patients with muscular dystrophy to have genetically confirmed X-linked EDMD, for a frequency of 0.54% of patients referred with a diagnosis of LGMD.e10
Transmembrane protein 43 (TMEM43) encoding LUMA/EDMD5.
A Class III study of 41 patients with the EDMD phenotype identified 2 patients with the heterozygous missense mutations p.Glu85Lys and p.Ile91Val in TMEM43.e71
Myofilbrillar myopathies.
The term myofibrillar myopathy (MFM) refers to a group of myopathies characterized by the following specific histologic features: (1) amorphous, hyaline, or granular material in the muscle fibers on trichrome-stained sections; (2) decreased oxidative enzyme activity in many abnormal fiber regions; (3) congophilia of the hyaline structures; (4) small rimmed vacuoles; and (5) myofibrillar degeneration on electron microscopy (EM).e51 The disorder is genetically heterogeneous. In this section we discuss all MFMs with identified genetic defects.
Myotilin (also LGMD1A).
One Class I studye5 and 3 Class III studiese8-e10 were reviewed. In a Class I population study of patients with muscular dystrophy in northern England, 1,105 cases registered and followed by the neuromuscular team at the Institute of Human Genetics, Newcastle University were studied. Diagnoses were obtained in 836 patients (75.7%). The combined prevalence of inherited myopathies was 37/100,000. LGMD comprised the fifth major category, with 68/1,105 cases, or 6.15%. No cases of LGMD1A were identified. However, 2 patients diagnosed with MFM had a mutation in the myotilin gene. This corresponds to a frequency of 0.18% (95% CI 0–0.4) of the clinic population, for a point prevalence in the population of 0.07 (95% CI 0–0.2) per 100,000.e5 In a Class III study, 6/57 (10.5%) families with MFM were found to carry myotilin mutations.e9 A large multicenter Class III study enrolled 370 patients with LGMD from 337 families. Genotype analysis was directed by the phenotype and muscle biopsy protein abnormalities. Of 297 patients, one was found to have myotilinopathy, for a frequency of <1%. However, because only 179/297 patients had undergone mutation analysis at the time of publication, it is possible that this number is an underestimate.e10 In another Class III study, 44 families with LGMD1, 14 with LGMD2, 24 with facioscapulohumeral dystrophy, 2 with scapuloperoneal dystrophy, and 2 with unclassified autosomal dominant dystrophies were screened for myotilin gene mutations. A myotilin gene mutation was found in one Argentinian family, for a frequency of 1/58 families with LGMD (1.7%).e8
Desmin (also LGMD1E).
One Class I studye5 and one Class III studye51 were reviewed. The Class I study reported the prevalence of desminopathy to be 0.17/100,000 (95% CI 0–0.3).e5 In the Class III study, desminopathy was seen in 4/63 (6.3%) unrelated patients with MFM.e51
αB-Crystallin.
One Class III study was reviewed.e51 Two of 63 patients (3%) with MFM were found to carry a mutation in CRYAB.
Z-band alternatively spliced PDZ motif-containing protein (ZASP) (also known as Markesbery Griggs distal myopathy).
One Class III study was reviewed.e72 Among 54 unrelated MFM patients without mutations in desmin, αB-crystallin, or myotilin, 11 patients (20.3%) were found to carry a mutation in LDB3, the gene that encodes ZASP.
BCL2-associated athanogene 3 (BAG3).
One Class III study was reviewed.e73 Among 53 unrelated MFM patients without mutations in desmin, αB-crystallin, myotilin, ZASP, or filamin C, 3 patients (5.6%) were found to carry a mutation in BAG3.
Autosomal recessive hIBM/Nonaka myopathy.
Glucosamine (UDP-N-acetyl)-2-epimerase/Nacetylmannosamine kinase (GNE) quadriceps-sparing myopathy. Two Class III studies were reviewed.e10,e74 A large multicenter Class III study enrolled 370 patients with LGMD from 337 families. Genotype analysis was directed by the phenotype and muscle biopsy protein abnormalities. One of 297 patients was found to have a mutation in the GNE gene, for a frequency of <1%. However, because only 179/297 patients had undergone mutation analysis at the time of publication, it is possible that this number is an underestimate.e10 In another Class III study, 92 of 1,000 (9.2%) Persian Jewish volunteers in Israel demonstrated heterozygous mutations in the GNE gene and therefore carrier status for hIBM.e74
Clinical Questions 2, 3, and 4 are addressed together in the following section because they evaluate different aspects of the phenotype of LGMD: 2. In patients with muscular dystrophy, what is the association between specific clinical features, degree of CK elevation, and subtypes of these disorders? 3. In patients with LGMD or distal muscular dystrophy, what is the association between specific muscle biopsy features and subtypes of these disorders, in particular rimmed vacuoles, inflammation, and inclusions? 4. How often do patients with muscular dystrophy and its specific subtypes have significant respiratory abnormalities (FVC <50% predicted), cardiac abnormalities (EF <50%, evidence of hypertrophic cardiomyopathy or generalized wall motion abnormality, arrhythmias, conduction defects), or bone loss (osteoporosis or bone mineral density 2.5 SD below peak bone mass, osteopenia or bone mass of 1.0–2.5 SD below peak bone mass)?
We did not include EDMD3 and EDMD4 due to SYNE1/nesprin-1 and SYNE2/nesprin-2 mutations because there was not enough evidence for a detailed assessment of phenotypes. No studies were available evaluating bone loss in LGMD.
LGMD1A (myotilin).
This is discussed in the section on MFM.
LGMD1B (lamin A/C).
There were 47 Class III studies.e12,e15,e16,e75-e118 Mutations in the LMNA gene result in diverse phenotypes, including LGMD1B, AD-EDMD, dilated cardiomyopathy with conduction system disease, Dunnigan type familial partial lipodystrophy, mandibuloacral dysplasia, Hutchinson-Gilford progeria syndrome, restrictive dermopathy, and a form of dominant-intermediate Charcot-Marie-Tooth syndrome (CMT). Phenotypic variability has been reported even in kinships with the same mutation.e95,e96,e103 Onset was congenital to adult life (fifth decade) with humeroperoneal (AD-EDMD) and limb-girdle phenotypes. The AD-EDMD phenotype was characterized by proximal muscle weakness in the upper extremities with preferential involvement of humeral muscles, both proximal and distal weakness in the lower extremities, elbow and ankle contractures, and spine rigidity (cervical > thoracic and lumbar). Of note, in some series contractures were seen only late in the disease course or not at all, which is different than X-linked EDMD, in which contractures are invariably present early in the disease course.e75,e81,e88,e106,e116,e118 Occasional patients had pseudohypertrophy of the calves or scapular muscles. Atrophy was appreciated in humeral muscles (biceps > triceps) and below the knees, particularly in the medial gastrocnemius.e75 Scoliosis of the thoracic spine was also noted in addition to rigidity. Scapular winging was uniformly seen and was correlated with the severity of weakness.e75
The LGMD phenotype was characterized by proximal leg-greater-than-arm weakness, but often with preferential involvement of humeral muscles. Again, flexion contractures of elbows and Achilles tendons were usually minimal or developed late in the course.e106,e116,e118 Cardiac abnormalities (arrhythmias, conduction defects, and dilated cardiomyopathy) were common and may be the only presenting feature of laminopathy. Pacemakers or intracardiac defibrillators were commonly implanted because of arrhythmias and the risk of sudden cardiac death. Cardioembolic stroke occurred because of associated arrhythmias. Many patients also required cardiac transplantation because of congestive heart failure (CHF) from dilated cardiomyopathy.
CK levels were normal or slightly elevated—most series had a CK average of <5 times the upper limit of normal (ULN). EMG usually revealed nonspecific myopathic features. MRI/CT demonstrated fatty infiltration in the posterior compartment of the thigh and calves.e80 The medial gastrocnemius and soleus appeared to be preferentially involved in laminopathies in some studies, even in patients who had only cardiac abnormalities without muscle weakness clinically.e77,e84,e85 However, other studies have found both the lateral and the medial gastrocnemius to be involved equally.e80 In both AD-EDMD and LGMD1B phenotypes, other muscles that were involved included the glutei, quadriceps, adductor magnus, and hamstrings.e77,e106 One study attempting to differentiate AD-EDMD and Bethlem/Ullrich myopathies (collagen VI disorders), which can also be associated with contractures, found that the quadriceps were relatively spared and the hamstrings were more severely involved in ADEDMD.e80 Muscle biopsies revealed nonspecific myopathic features (e.g., variability in fiber size with or without necrotic and regenerating muscle fibers and increased endomysial connective tissue). One series reported endomysial inflammation in young children who were initially felt to have an inflammatory myopathy.e106 Biopsies have shown normal or reduced immunostaining for lamin A/Ce79,e81,e83 and laminin beta-1.e90 Rare rimmed vacuoles were reported.e78 On EM, nuclear alterations in about 10% of the preserved muscle fibers with peripheral heterochromatin loss or detachment from the nuclear envelope and interchromatin texture alterations have been reported.e113
As mentioned, mutations involving lamin A/C have also been associated with dominantintermediate CMT (not reviewed in this manuscript),e119 and some patients have signs of both a myopathy and a neuropathy on EMG and muscle biopsy.e93
LGMD1C (caveolin-3).
Thirteen Class III studies were reviewed.e120-e132 Patients have been reported from the United Kingdom, Italy, Spain, Sweden, and Japan.e120-e122,e130,e132 Age at symptom onset ranged from 5–81 years. The clinical phenotypes varied. Intrafamilial variation was not uncommon, and patterns of involvement included proximal arm and leg weakness, distal weakness, rippling muscle disease, or asymptomatic hyperCKemia. Prominent muscle cramps (spontaneous, postexercise, or myalgias) were noted in most patients.e120-e122 In 3 studies, distal hand weakness and atrophy was noted in 9/23 patients.e121,e122,e132 In one series, 3 of 10 patients presented with toe walking but did not have distal leg weakness.e121 Rippling muscle disease was the sole manifestation of caveolinopathies in several families. One series reported generalized percussion-induced rapid contractions (PIRCs) in the face, neck, and extremities as a constant feature, but actual rippling muscles were seen less frequently (12/19).e131 In a study of 23 patients from a large Swedish family, all except 2 presented with muscle stiffness.e130 Percussion-induced muscle mounding and PIRCs were noted in all 23 patients, muscle rippling in 12, calf hypertrophy in 9, and generalized hypertrophy in 2, whereas weakness was not observed. Rhabdomyolysis and myoglobinuria were rarely noted (1/10 patients in one study, 4/19 in another).e121,e131 Most patients, even when asymptomatic, exhibited calf hypertrophy.e120,e121 Six of 7 in one family had pes cavus.e122 Scapular winging, facial weakness, and rigid spine were not seen. Mild contractures were seen in one study (finger flexor and hamstrings in 2/10 patients).e121
CK levels were elevated 3–30 times above normal.e120-e122,e130 Four of 7 patients in one family had asymptomatic hyperCKemia (2 patients subsequently developed weakness in the hands 2 decades later).e122 Rare patients with cardiac involvement have been described, although clinical cardiac manifestations are distinctly uncommon.e123,e124 Muscle biopsies were normal or mildly myopathic or dystrophic, without specific diagnostic features.e121 Reduced staining for caveolin-3 on the sarcolemma on immunohistochemistry has been reported.e121,e122,e130,e132
LGMD1D (DNAJB6).
The nomenclature of LGMD1D and LGMD1E has been confusing in the literature. AD LGMD linked to chromosome 7q36 has been termed LGMD1D as well as LGMD1E. In this guideline, we refer to 7q36-linked LGMD with mutations in the DNAJB6 protein as LGMD1D. Three Class III studies were reviewed.e133-e135 Two studies describe the same Finnish families and are reviewed together.e133,e134 One of the studiese134 also includes 2 Italian families and 2 US families, whereas the thirde135 describes 2 families from the US (ethnicity not mentioned). Disease onset was in the third to sixth decade, except for 2 US patients from different families with onset at ages 14 and 18 years. All patients had moderate to severe proximal muscle weakness in the lower extremities; often the quadriceps was relatively preserved compared to the hamstrings.e135 Proximal upper extremity weakness was absent or milder than lower extremity weakness. In one study, distal lower extremity weakness involving the posterior compartment more than the anterior compartment was noted in all 9 families.e134Another study reported one family in which 3 affected members had distal muscle atrophy and weakness in the legs as well as the arms and mild to moderate proximal weakness.e135 Some patients had heel cord contractures. Cardiorespiratory involvement was notably absent.
Serum CK levels ranged from normal to 10-fold elevated but averaged about 2–3 times the ULN in most cases. Muscle biopsy in all studies revealed a myopathy with rimmed vacuoles, features suggestive of an MFM in 7/9 families.e134
LGMD1E (desmin).
This is discussed in the section on MFM.
LGMD2A (calpain-3).
One Class Ie136 and 36 Class IIIe17,e19,e22-e26,e28,e30,e33,e34,e85,e137-e160 studies were reviewed. Most cases had onset between 5 and 20 years of age. The mean age at onset across studies spanned 9.8–21.8 years,e138,e149 but the range was broad, from 2–65 years.e30,e33 Approximately 20%–50% of patients eventually became wheelchair dependent,e136,e137,e140,e148 and the mean time from disease onset to loss of ambulation ranged from 9.4–23.6 years.e140,e148 Onset of weakness occurred in the lower extremities alone in 80%, in the lower and upper extremities in 13%, in the upper extremities alone in 3%, and with isolated hyperCKemia in 4%.e25,e148,e150 Hip extensor, hip adductor, and knee flexor muscles were most affected.e33,e148,e149,e152,e157,e158 Facial weakness was uncommon; it was reported in less than 5% of cases (4/96 cases).e25,e138,e148,e150,e152 Calf hypertrophy was seen in 51/126 (40%) cases.e23-e25,e33,e148,e150,e157 However, in the Class I study, calf hypertrophy was described as rare. e-136 Scapular winging often was not present at diagnosis but over time became nearly universal. Overall, scapular winging was present in more than 80% of patients (78/95).e24,e33,e34, e150,e152,e157 Dysphagia was not present in any of 51 patients across 3 studies.e138,e148,e157 Likewise, dysarthria and hoarseness of voice were seen in none of 20 patients.e148 Contractures occurred in 25% (18/71) of patients and predominantly affected the ankles.e17,e30,e33,e34 There was essentially no symptomatic cardiac involvement. Across all studies, 9% (17/198) of patients had abnormalities on cardiac testing. In a Class I study, 7/35 patients had ECG abnormalities.e136 The abnormalities included nonspecific conduction abnormalities in 5/35 patients and repolarization abnormalities in 2/35 patients. No significant abnormalities were found in the 29 patients who underwent echocardiography.e136 None of the patients in the Class I study had cardiac symptoms. Eight Class III studies evaluated cardiac testing.e17,e25,e138,e148,e150,e152,e156,e157 Five percent (8/163) of patients had ECG changes, including premature atrial or ventricular beats, ST segment elevations, atrial fibrillation, atrioventricular conduction block, or bundle branch blocke25,e150,e156; 2% (4/163) had abnormalities on echocardiography, including mild anterior cardiac wall dysfunction, mildly impaired left ventricular function, slight diminution of left ventricular contractility, and a left ventricular ejection fraction (LVEF) slightly below 50%.e25,e148,e150,e156 Four Class III studies found no abnormalities on ECG or echocardiography.e17,e138,e152,e157 Significant respiratory involvement was very infrequent until late in the disease course. Seven percent (8/117) of patients had a restrictive pattern over multiple studies,e17,e136,e138,e150,e156,e158 but 12/20 had an FVC reduced to 30%–50% of normal late in the disease course.e148 Cognitive dysfunction was not reported (0/75 patients).e17,e25,e148,e157
MRI demonstrated fatty and fibrous replacement in the gluteal, hamstring, adductor, soleus, and medial gastrocnemius muscles.e85,e144,e149,e152,e155 CK levels were most often more than 10 times the ULN, with a mean of 19 times the ULN and a range of normal to 110 times the ULN. CK levels were elevated >10 times the ULN in 90/146 (62%), 2–10 times the ULN in 50/146 (34%), and were normal in 6/146 (4%).e19,e22,e24,e25,e30,e33,e34,e136,e142,e152,e153,e157,e158 On muscle biopsy, lobulated fibers were frequently seen: 33/47 biopsies (70%) on NADH-stained sections.e25,e26,e148 Rimmed vacuoles and inclusions were not features, but inflammation, including eosinophils, may be seen on some biopsies.e30,e140,e150,e160 Western blot analysis of muscle calpain-3 in patients with LGMD2A can show a total or partial deficiency or no deficiency. Furthermore, calpain-3 may be reduced in muscle from patients with LGMD2B and patients with LGMD2J, whereas dysferlin immunostaining may be reduced on muscle biopsies from patients with LGMD2A.e150 Thus, genetic testing is required to confirm all cases.
LGMD2B/Miyoshi myopathy (dysferlin).
One Class II studye161 and 54 Class III studiese17,e23,e24,e137,e145,e147,e149,e155,e162-e207 were reviewed. The mean age at onset has been reported as 18.4–31.9 years across reports, with most studies falling between 19 and 23 years. The range for age at onset was 3–60 years, with most studies describing a range of 10–35 years.e17,e23,e24,e145,e147,e149,e162-e164,e166-e168,e170,e171,e173- e176,e178,e182,e183,e185,e188-e191,e193,e195,e197,e198,e200-e205, e169,e192,e200,e206
The dysferlinopathies involve 2 principal clinical phenotypes that can merge over time.e167,e168 The Miyoshi phenotype is characterized by distally predominant weakness principally involving the posterior compartment (calf), as described in 17/19 dysferlinopathies,e162 14/29,e163 18/25,e166 21/26,e147 9/14,e167 11/36,e168 4/8,e177 2/9,e178 46%,e188 and in several other series.e165,e170,e172,e174-e176 The second phenotype is characterized by a limbgirdle pattern of weakness, accounting for 15/29 dysferlinopathies,e163 3/25,e166 8%,e147 7/9,e178 23/33 (70%),e23 22/36 (61%),e168 40%,e188 “most” of 37,e17 and 5/14e167 in various series. There can be preferential weakness of the biceps in the arms followed by lesser weakness of the deltoid and tricepse149,e163,e167,e173,e205; the weak biceps and relatively preserved deltoid can produce a “deltoid bulge.”e173 Even in patients presenting with a “limb-girdle” phenotype, the gastrocnemius muscle was still notably atrophic, particularly the medial aspect.e23,e167,e168 Rare patients presented with anterior leg (tibialis anterior) weakness and foot drop, occurring in 3/8,e177 2/19,e162 2/30,e188 4 cases in one family,e207 and 2/11e203cases. Weakness was commonly asymmetric.e167,e168 One study described a “diamond on the quadriceps” bulge, affecting 21/31 patients with dysferlinopathy who presented with both the Miyoshi and limb-girdle pattern of weakness.e170 Calf atrophy was typical,e189,e195 and atrophy of the anterior shin was sometimes seen.e195 Calf hypertrophy and pain have been reported early in the course in 5/29,e163 1/3,e24 3/14,e167 5/39,e23 and 6/21e173 patients with dysferlinopathy. Partial atrophy of the biceps has been observed in both the Miyoshi and limb-girdle phenotypes,e189 and selective atrophy of the shoulder girdle muscles producing a “double calf’s head on a trophy” sign has been described.e195 Scapular winging, dysphagia, dysarthria, and contractures were not reported. Some dysferlinopathy patients can present with asymptomatic hyperCKemia or recurrent myoglobinuriae192; symptomatic carriers have also been described.
Respiratory and cognitive dysfunction have not been described. Cardiac involvement was uncommon.e96,e162,e168,e178,e189,e191,e201,e205 Nonspecific ECG changes have been reported in some patients.e17,e161 Echocardiography has demonstrated left ventricular hypertrophy or reduced EF in rare patients.e17,e161,e204 Subclinical cardiac fibrosis or diastolic dysfunction was reported in a few patients by cardiac MRI.e197 Cardiac muscle biopsies in 2 patients revealed absence of dysferlin from the sarcolemma with perivascular and interstitial fibrosis.e204
Muscle CTe175,e193 and MRIe149,e155,e165,e167,e172,e189,e191,e203 reported preferential involvement of the posterior compartments of the distal and proximal legs. By MRI, the pattern of muscle involvement appeared similar for patients with both the Miyoshi and limb-girdle phenotypes,e189 with early involvement of the gastrocnemius and thigh adductors. CK levels were typically markedly elevated,e17,e162,e165,e167,e168,e171,e172,e173,e175,e177,e178,e182,e183,e185,e189,e191,e193,e200,e201,e204,e205,e206 up to 10–30 times,e163 23–40 times,e24 36 times,e23 20–100 times,e174 or 15–30 timese185 the ULN. Muscle biopsies were characterized by dystrophic changes.e145,e162- e167,e169,e171,e173,e175,e177,e178,e181,e183,e184,e193,e201,e202,e205-e207 Perivascular and/or endomysial inflammatory infiltrates were common.e162,e164,e166,e169,e171,e173,e181,e183,e184,e187-e189,e193,e201,e202 Amyloid deposits were detected by Congo red staining in blood vessel walls and in perimysial connective tissue in one studye173; 4/6 specimens in a second study also contained sarcolemmal and interstitial amyloid deposits in skeletal muscle.e200 Absence or reduction of dysferlin by immunofluorescence and/or Western blot staining was characteristic.e17,e147,e166-e169,e176,e177,e182,e183,e192,e193,e196,e198,e200-e202 Rimmed vacuoles and inclusions were not a common feature, although a single study described rimmed vacuoles in 4/14 patients with a Miyoshi phenotype.e188
LGMD2C (γ-sarcoglycan).
Two Class Ie208,e209 and 15 Class IIIe17,e23,e41,e43,e210-e220 studies were reviewed. This dystrophy occurs worldwide but may be more common in Roma/Gypsye209,e216 and Tunisian populations.e208,e220 Onset occurred in the early childhood to adult years, but most series had an age at onset in early childhood, with a range of 1–13 years (mean 6.1 years) in one Class I studye208 and a range of 2–8 years in the other Class I study.e209 However, other series had slightly later ages of onset, ranging from 2–23 years (mean of ~11 years).e17,e23 Patients presented with proximal leg weakness greater than proximal arm weakness. Scapular winging, calf hypertrophy, macroglossia, ankle contractures, and scoliosis were common, at least in the Roma populations with the disorder.e209,e216 Age at loss of ambulation ranged from 11–37 years (mean 16 years) in one Class I studye208; 81% of patients were wheelchair dependent by age 14 years in the other Class I study.e209 Normal intelligence was noted in 2 small series.e41,e215 The largest series of 68 patients with LGMD2C found no patient with clinically relevant cardiomyopathy.e216 Most small studies reported that patients had normal ECG and echocardiography.e41,e210,e218 In one study, ECG and echocardiogram were normal in 2/3 patients and revealed abnormal contraction of the interventricular septum in 1/3e217; 4/10 patients had dilated cardiomyopathy in another study.e211 Ventilatory muscle weakness requiring noninvasive ventilation developed in 2/5,e217 but respiratory function was normal in other small series.e43,e215 CK levels were elevated 4–100 times normal in most series.e17,e23,e208,e211-e213,e216 Muscle biopsies revealed markedly reduced or absent γ-sarcoglycan on immunohistochemistry, whereas immunohistochemistry of other sarcoglycans was more variable (normal or moderately reduced).e23,e208,e210,e211,e212,e216,e219
LGMD2D (α-sarcoglycan).
Eighteen Class III studies were eviewed.e17,e19,e23,e43,e145,e149,e156,e164,e213,e218,e219,e221-e227 LGMD2D has been described in French, Italian, Moroccan, Algerian, Finnish, German, white Brazilian, and African-Brazilian families.e19,e23,e221,e223,e225,e226 Symptom onset occurred at 1–30 years of age (mean 10.5 years). The legs were weaker than the arms. The glutei and hip adductors were involved more than the psoas and the thigh muscles; the quadriceps and hamstrings were involved equally. Distal lower extremity weakness was minimal and, if present, involved the tibialis anterior. In the upper extremity, the deltoids, serratus anterior, trapezius, and latissimus dorsi and rhomboids were involved early. The infraspinatus was affected more than the supraspinatus, and the biceps was involved but less so than the infraspinatus and supraspinatus; the triceps, pronators, and supinator were spared. Trunk extensors were involved but neck flexors only minimally so. Scapular winging, thigh atrophy, and calf hypertrophy were seen in the majority of patients across studies.e213,e221,e223 The weakness varied widely in severity, and intra-/interfamilial variation was common. In one study, 4/24 patients lost ambulation before age 16, whereas 9 were ambulatory, 3 of whom were aged 50 or older (age range 6–56 years, mean 34.4 years, although the 2 youngest patients had been followed only 1 and 3 years).e213 Intellectual development was noted to be normal in all 12 cases in one study.e226 CK levels were elevated in all patients but varied widely from 2–100 times normal.e17,e19,e23,e213,e221,e223,e227
Symptomatic cardiomyopathy was not common, at least early on. ECG revealed nonspecific abnormalities in a minority of patients.e156 Echocardiography was usually normal,e156,e213 although a minority had findings of a dilated cardiomyopathy.e218,e226
Severe ventilatory muscle weakness has been reported in up to one-third of patients.e43,e213 CT scan of the lower limbs revealed early involvement of pelvic muscles, especially the glutei and posterior and deep anterior thigh muscles.e221 The medial femoral muscles were spared in the mildly affected patients. Hypertrophy of rectus femoris, sartorius, and gracilis was observed in a mildly affected patient. Distal muscles were spared except in the severe cases, when the tibialis anterior was involved. Muscle MRI in 2 patients demonstrated more severe involvement of the quadriceps than the posterior thigh muscles, with hypertrophy of the gracilis and sartorius; one of these patients also had changes in the soleus, gastrocnemius, and peroneus longus.e149 Muscle biopsies revealed variable immunohistochemistry staining of the sarcoglycan complex.e219,e225,e226
LGMD2E (β-sarcoglycan).
Thirteen Class III studies were reviewed.e17,e23,e24,e40,e43,e144,e156,e164,e213,e219,e228-e230 β-Sarcoglycanopathy was reported in patients of Turkish, Italian, East Indian, Tunisian, white Brazilian, and African-Brazilian descent.e17,e23,e24,e40,e228,e229 The age at onset was 8–20 years (mean 6.9 years).e17,e40,e213 Clinical features varied, including mild proximal muscle weakness (pelvic > shoulder girdle) in 6/12 patients and symptomatic hyperCKemia with calf hypertrophy in 2/12 patients in one study, but a severe DMD-like picture was noted in 30%–70% of cases across reports.e23,e24,e213,e228 Loss of ambulation and wheelchair dependence was variable and seen in childhood or early adulthood.e23,e40,e229 Patients may have calf hypertrophye24,e213,e228 or scapular winging.e24 Spinal scoliosis and Achilles contractures were rare.e229 The type and severity of cardiac involvement varied across studies. Normal cardiac function was reported in some studies.e24,e213,e228 However, cardiomyopathy was apparent by echocardiogram in some studies.e40,e43,e230 One patient died at age 21 of cardiac arrest in one series.e23 Respiratory function was stated to be normal in all 12 patients in the Turkish study, although details were not mentioned.e228 However, some patients developed ventilatory failure over time.e43,e156,e213
CK levels were elevated 2–110 times normal (mean 14 times) across studies.e17,e23,e213 Muscle imaging in one patient revealed severe fatty atrophy of the shoulder and pelvic girdle.e144 Muscle biopsies revealed typical dystrophic features along with variable immunohistochemical staining to the sarcoglycans. A complete absence of immunohistochemical staining tended to be associated with earlier onset in one studye17 and more severe phenotype in 2 studies.e213,e219
LGMD2F (δ-sarcoglycan).
Eight Class III studies were reviewed.e17,e23,e40,e43,e44,e213,e219,e231 Two of these studies had only one patient with LGMD2F each because they studied sarcoglycanopathies as a whole, but they were included because of the rarity of the disorder.e17,e213 The Brazilian studiese23,e43,e219,e231 appear to describe the same patients, with a few additional patients in the later papers, and are reviewed together. The ethnicity of the described patients was East Indian, white Brazilian, and African-Brazilian.e23,e40 The age at onset was 4–10 years.e17,e23,e213,e231 Eight of 9 patients had a severe DMD-like presentation with onset in early childhood and wheelchair dependence between 11 and 16 years. One patient was ambulatory for short distances at 19 years.e23,e231 Two patients, one aged 13 years and the other aged 17 years, also had a DMD-like presentation with cardiac involvement (one presymptomatic cardiomyopathy and pulmonary hypertension, one dilated cardiomyopathy). Respiratory involvement was noted in both patients (one mild, FVC 76%; one moderate, FVC 54%).e43 CK levels were elevated 5–24 times normal, with a 100-fold elevation in one case.e23,e40,e43,e213 Immunohistochemical analysis revealed total absence of all sarcoglycans in 3 patients and absence of α-, β-, and δ-sarcoglycan with partial deficiency of γ-sarcoglycan staining in one patient.e44,e219
LGMD2G (telethonin).
Three Class III studies were reviewed.e23,e232,e233 The family described in one of the studiese23 was also included in another study.e232 The initial description by the same authors was cross-referenced to obtain details of phenotype. Twelve patients from 3 Brazilian families were described. Onset was between 2 and 15 years. Patients had lower extremity distalgreater-than-proximal or proximal-greater-than-distal weakness. In the upper extremities, the weakness was more proximal than distal. Progression of the disease varied within families. Calf hypertrophy was seen in all affected patients of one family. CK levels were elevated from <10 to 30 times normal. Cardiac involvement was noted in 3/6 affected members of one family; the type of involvement was not specified. Muscle biopsy was remarkable for abundant rimmed vacuoles in 2 families.e233
LGMD2H/TRIM32/sarcotubular myopathy.
Four Class III studies were reviewed.e234-e237 The initial families were of Hutterite descent.e234,e237 The age at onset ranged from birth to the seventh decade of life. Most affected individuals had a limb-girdle pattern of weakness. Facial weakness, scapular winging, calf hypertrophy, Achilles contractures, neck flexor weakness, and exercise-induced myalgias were noted in a few patients (2/5,e234 2/4,e235 1/4e236); neck flexors were weak and tendo Achilles contractures were noted in 2/4 patients.e234-e236 Peripheral neuropathy with slowed nerve conduction velocity was seen in 3/5 patients in one study.e235 ECG in 9 patients showed right bundle branch block in 2.e235,e236 Respiratory function was normal in 1/4 patients and FVC was reduced to 41% in 1/4 patients.e236
CK levels were normal to 20-fold elevated. The characteristic muscle biopsy feature was many small vacuoles more prominent in type II fibers, although in isolated cases type I fibers had more vacuoles. On EM, the smallest vacuoles were focal dilations of the sarcoplasmic reticulum and coalesced to form larger vacuoles, often with degeneration of their muscle membranes.e234,e235
LGMD2I/fukutin-related protein (FKRP).
Three Class I studies,e57,e69,e238 one Class II study,e239 and 27 Class III studiese17,e52-e56,e144,e149,e197,e240-e257 were reviewed. LGMD2I is a common cause of LGMD in white populations in northern Europe, Denmark,e69 Italy,e17 Germany,e52,e257 Norway,e57 and the United Kingdom,e245 and has also been described in North America, Brazil,e238 and in families of Tunisian and Bedouin descent. The mean age at onset was 12.7 years in one Class I studye57 and 20.2 years in another Class I study.e238 In Class III studies the age at onset ranged from 1.5–54 years, with means ranging from 9–23.2 years.e17,e52-e54,e149,e240,e242, e245,e256,e257 Weakness was symmetric and proximal, affecting the legs earlier and more severely than the arms, as reported in 2 Class I studiese69,e238 and 11 Class III studies.e52-e54,e56,e149,e240,e242,e243,e245,e253,e257 In the legs, hip flexion and hip adduction were particularly affected,e69,e149,e240 whereas in the arms shoulder adductione149,e240 and elbow flexione69,e149 were especially affected. Facial weakness was reported in 3/18 in one Class III studye256 but was not reported in one Class I study and 3 Class III studies.e56,e69,e240,e257 No dysarthria or dysphagia was reported, nor atrophy of any specific muscle groups. Scapular winging was noted in 3/20 patients,e53 6/11 patients,e240 and 3/7 patients.e56 Contractures did occur but were not a prominent feature; occasional ankle contractures were reported in 2/7,e56 2/16,e245 and 2/27 homozygotes and in 5/11 compound heterozygotes (Class I).e69 One Class III study reported contractures of the tibialis anterior (17/18), hips (3/18), knees (4/18), and elbows (3/18),e256 whereas no contractures were observed in other series.e240,e242,e243,e257 Calf hypertrophy was common.e52-e56,e69,e238,e242,e245,e246,e249,e256,e257 Hypertrophy was also noted in some patients in the brachioradialis,e54,e245 the thigh,e256 and the tongue.e53-e55,e69,e240,e245,e246,e257 Dilated cardiomyopathy was common clinically or by echocardiogram.e52,e53,e55,e69,e197,e239,e240,e245-e249,e252,e253,e255-e257, e197 One study using cardiac MRI disclosed myocardial fibrosis in 4/7. In this study, age, muscle strength, ability to ambulate, severity of dystrophic changes on muscle biopsy, and age at symptom onset did not correlate with cardiac involvement. Respiratory dysfunction was common, with a reduced FVC in a restrictive pattern observed in 2 Class I studiese57,e69 and in 7 Class III studies.e17,e53,e55,e56,e240,e243,e245 FVC was reduced 45%/62%/66%/82%/50% of the time,e53,e240,e243,e245,e253 often moderately to severely; respiratory support in the form of noninvasive positive pressure ventilation or assisted ventilation was necessary in 20% to 25% of patients in the Class I studiese57,e69 and in up to 45% of patients in the Class III studies.e53,e55,e57,e156,e240,e243,e245,e256 Cognitive dysfunction was not mentioned in most studies, but was specifically noted to be absent in 2.e53,e242 Formal cognitive testing was performed in 2 studies.e240,e253 In one, 10/11 patients had normal verbal/written memory, and only 1/11 had a low IQ.e240 In the second, mild impairment of executive function and visuospatial planning without a global reduction in IQ was common.e253
CK levels were almost always more than 10-fold elevated, as reported in 2 Class I studiese69,e238 and 12 Class III studies.e17,e52-e55,e240,e242-e245,e251,e257 Episodes of myoglobinuria were reported in 5/14 in one Class III studye250 and in 7/26 in another.e253 MRI studies of the legs revealed abnormal signal and fatty infiltration of the psoas, gluteus maximus, and thigh adductors, with relative preservation of the anterior thigh.e144,e149,e240 The gracilis and sartorius muscles were involved later and were sometimes spared or even hypertrophied.e149 MRI studies of the arms revealed abnormalities in the serratus, subscapularis, infraspinatus, and supraspinatus, with relative preservation of the triceps; the deltoids and biceps were either spared or involved later. Muscle biopsies were notable for dystrophic changes, including necrosis, evidence of degeneration and regeneration, variation in fiber size, internal nuclei, and fibrosis.e56,e69,e240,e245 Reduced α-dystroglycan immunoreactivity was observed.e54,e241,e244,e257 Rimmed vacuoles, inclusions, and inflammation were absent.
LGMD2J/Udd distal myopathy/hereditary myopathy with early respiratory failure (titin).
Ten Class III studies were reviewed.e258-e267 An additional article referenced in Udde258 was also reviewed for clinical details.e268 There are 3 major clinical phenotypes of titinopathies: autosomal recessive LGMD2J, autosomal dominant distal myopathy, and autosomal dominant hereditary myopathy with early respiratory failure (HMERF).
LGMD2J.
LGMD2J has been reported mainly in Finnish and French populations.e258,e259,e264 Onset of weakness was in the first 3 decades of life, but one Finnish patient was noted to have initial delayed motor milestones that subsequently stabilized, only to develop weakness around age 10 years.e264 All patients had severe proximal muscle weakness and atrophy involving the pelvifemoral and scapulohumeral muscles, with milder distal weakness (anterior tibial, gastrocnemius, forearm, and hand), and developed severe generalized weakness and wheelchair dependence over the subsequent 20 years. The face was spared, and scapular winging was described in only one patient. Muscle hypertrophy was not noted. One Finnish patient with onset in the early school years died at age 64 years from respiratory failure; no details were provided. At autopsy the heart was normal without signs of heart failure. One of the 7 patients initially described had atrial fibrillation, and another patient had “occasional cardiac arrhythmia.” Echocardiogram performed in 3/7 patients was normal. In contrast to this presentation, an earlyonset recessive myopathy with severe cardiomyopathy characterized by delayed milestones and predominantly lower extremity proximal and distal weakness (but also involving proximal upper extremity, trunk, and face weakness and ptosis) has been described in Moroccan and Sudanese patients. In these 5 patients, pseudohypertrophy of the thighs and calves contrasted with atrophy of the upper limbs. Spinal rigidity and moderate joint contractures appeared in the first decade. The muscle disease was mild, but a progressive, severe dilated cardiomyopathy developed in all 5 patients, with rhythm disturbances. Sudden death occurred in 2 at 19.5 and 17.5 years.e267 CK elevation was usually moderate (3–5 times normal); one patient with >10-fold elevation was reported. Muscle MRI was abnormal in all 22 patients in one study. Eight of the 22 patients had fatty replacement of leg muscles (anterior compartment only in 6, both anterior and posterior compartments in 2). Fourteen patients had both thigh and leg involvement. The hamstrings were uniformly involved: quadriceps in only 3 patients, gracilis in one patient. The lateral leg compartment was usually spared, being involved in only one patient at a late stage of the disease.e263 Muscle biopsies revealed dystrophic features, and rimmed vacuoles were usually absent or rare.e258,e264,e268
Udd distal myopathy.
Distal myopathy due to titin mutations has been described in Finnish, French, and Belgian populations.e259-e262,e264 In contrast to the LGMD phenotype due to the same protein defect, the age at onset of distal myopathy is in the fifth to seventh decade of life. The muscle weakness predominated in the anterior tibial leg compartment in all patients, with mild weakness of the pelvifemoral and gastrocnemius muscles in a few patients (6/41 and 2/41). Atrophy of the anterior tibial muscles was noted in 26/41 and of the gastrocnemius in 1/12 patients.e259,e260 The tibialis posterior and peroneus longus were also mildly weak in one patient.e262 CK levels were normal or mildly elevated (30%–64%).e260,e261,e263 One Belgian patient had CK levels >10 times normal.e262 Cardiac involvement was absent.e261 CT scan revealed fatty infiltration in the anterior tibial muscles in 7/9 patients and patchy involvement in the gastrocnemius and pelvic muscles in one patient each.e258 In one Belgian patient, fatty degeneration of the tibialis anterior and extensor digitorum longus muscles, and to a lesser extent the gluteus medius and minimus muscles, was noted.e262 Muscle biopsy revealed rimmed vacuoles in 28% of patients in the largest study.e260
Hereditary myopathy with early respiratory failure.
HMERF recently has been reported to be caused by mutations in titin in Swedishe265 and Englishe266 families. The phenotype merges with that of LGMD2J and Udd distal myopathy. Like Udd, it is inherited in an autosomal dominant fashion and has an early predilection for the anterior compartment of the distal lower extremity leading to progressive foot drop. However, it tends to affect patients earlier in adulthood (range 18–71 years) and may affect the proximal muscles (legs greater than arms), as seen in LGMD2J.
The majority of patients have prominent calf hypertrophy. Ventilatory muscle weakness gradually develops over time; however, cardiomyopathy is not seen. One report noted that the most commonly affected muscles on MRI were the semitendinosus (20 of 21 subjects), the peroneus longus (16/21), and the obturator externus (15/21).e266 In the other series, MRI revealed fatty replacement mainly of the iliopsoas, rectus abdominis, obturatorius, and gluteus minimus muscles. Severely affected muscles in the thighs were the semitendinosus, gracilis, sartorius, vastus lateralis, intermedius, and medialis muscles, whereas the adductor longus muscles were relatively spared. In the lower legs, there was fatty replacement predominantly in the anterior and lateral compartments. Muscle histopathologic features included rimmed vacuoles, eosinophilic inclusions, desmin deposits, and extensive myofibrillar lesions with marked Z-disk alterations on EM resembling those described in MFM; thus, titin mutations of the HMERF phenotype should be added to the differential diagnosis of MFM.
LGMD2K (protein-O-mannosyltransferase 1 or POMT1).
One Class III studye59 evaluated 3 patients with LGMD, ethnicity unspecified. Onset of disease was in infancy in 2 patients and at 3 years in one patient. Details of muscle involvement were not described. Muscle hypertrophy was noted in all 3 patients, but muscles involved were unspecified. Other phenotypes associated with POMT1 mutations in this study included WWS (1), MEB/FCMD (1), and CMD-MR (3). CK levels were elevated >10 times. All patients had microcephaly and intellectual disability (mental retardation). Brain MRI showed minimal white matter changes in one patient and normal results in 2 patients. This study also included one patient with LGMD2K described by the authors previously; the studies were therefore reviewed together.e244
LGMD2L (anoctamin-5).
Four Class III studies were reviewed.e61,e269-e271 Earlier reports of these families were also reviewed for clinical details.e272,e273 The disorder was initially reported in patients of French Canadian, Finnish, or Dutch descent, but subsequently in Australian, Spanish, Italian, German, and Afghan patients. Patients manifested with a limb-girdle pattern of weakness (LGMD2L) or with distal weakness resembling Miyoshi myopathy. The latter has been referred to as Miyoshi myopathy type III (MM3), but these phenotypes often overlap over time. Patients also presented for evaluation of hyperCKemia.e61,e269-e271 One study included 7 patients with LGMD2L and 5 patients with distal myopathy.e269 Another study reported long-term follow-up of 2 patients of Finnish descent with MM3 as reported in the first study.e270 A third study found 25 patients with ANO5 mutations out of a cohort of 101 patients with recessive LGMD, calf weakness, or hyperCKemia.e61 The final study reported 4 patients: one with LGMD, one with distal myopathy, one with hyperCKemia, and one with weakness and atrophy of the quadriceps and medial calves.e271 Age at onset ranged from 20–55 years (mean 34.4 years) in LGMD2L and 20–51 years (mean 36 years) in patients presenting with distal myopathy. Five of 7 patients (71%) with LGMD2L had weakness of the pelvic and scapular girdles. One patient also had mild weakness of the calf muscles and another of the tibialis anterior. One patient had atrophy and weakness of the right biceps brachii and right posterior thigh atrophy on examination initially, despite involvement of lower extremity muscles on MRI, and developed asymmetric hamstring weakness after 2 years.e61 Asymmetric atrophy of the biceps and quadriceps was noted in many patients across reports. Patients with distal myopathy had early calf weakness with difficulty in toe walking; the calf muscles were noted to be atrophic, often asymmetrically. However, some patients had calf hypertrophy early in the course before atrophy ensued. Atrophy or weakness of the quadriceps was noted in some patients with LGMD and some patients with distal myopathy.e269-e271 Atrophy was also appreciated later in the course in the biceps and pectorals.e270 CK levels were elevated 8- to >20-fold. Five patients across the studies had asymptomatic hyperCKemia noted in the fourth decade or later, although one had calf hypertrophy. Echocardiography, ECG, and Holter monitoring were normal in all the patients who were tested across studies.e61,e269-e271 No pulmonary abnormalities were noted in one study.e61 In 2 patients, muscle MRI showed atrophy and fat replacement of the long head of the biceps brachii. Muscles most involved in the legs were the medial gastrocnemius, adductor magnus, hamstrings, tensor fasciae latae, and to a lesser extent the quadriceps, often asymmetrically.e61,e273 In one of the 2 patients with distal myopathy who were followed long-term, initial muscle MRI at disease duration of 10 years showed fibrofatty degeneration in the gastrocnemius. Subsequent studies revealed similar changes in the soleus and biceps brachii. The other patient with distal myopathy also had asymmetric changes in the adductor magnus, vastus lateralis and intermedius, and tensor fasciae latae.e270 Muscle biopsies revealed myopathic/dystrophic changes. EM revealed multifocal disruption of the sarcolemmal membrane.e61,e270
LGMD2M (fukutin).
Three Class III studies were reviewed.e59,e244,e274 There were only 5 patients from 3 families reported across these studies. The disorder was described in one Israeli family in one study and in the child of Jewish and East Indian parents in another study. The ethnicities in the other cases were unspecified.e274 The onset of illness was from 4 months to 4 years of age. The phenotype was described as LGMD without intellectual disability (mental retardation) in 2 studies, with lower extremities more affected in 2/3 cases and upper extremities more affected in 1/3 cases. Muscle hypertrophy was described in 4/5 cases in one study, but the muscle group was not specified.e59 In another study, 2/3 patients had lateral calf hypertrophy.e274 Unspecified contractures were reported in 2/5 patients.e59 Cognitive function as defined by IQ was normal in all 5 patients, and brain MRI showed mild hydrocephalus in one patient and normal results in 2 patients. Increased weakness with a febrile illness was reported in 2 patients. All patients had CK levels more than 10 times normal. The muscle biopsy was significant for the presence of inflammation with macrophages, CD8+ lymphocytes, and major histocompatibility complex class I antigen upregulation in 2 cases.e59,e274
LGMD2N (protein-O-mannosyltransferase 2 or POMT2).
Two Class III studiese59,e244 and one Class IV studye275 were available. The Class IV study was retained because of the rarity of the disorder. One Class III study was excluded because it reported only patients with MEB and CMD, which are not addressed in this guideline.e244 The other 2 studies describe only one patient each with LGMD2N.e59,e275 One patient presented with developmental delay at 18 months and had an LGMD phenotype with learning difficulties at age 20 years.e59 Hypertrophy was mentioned but not described further. Cardiac evaluation revealed right bundle branch block. The second patient was found to have elevated CK levels incidentally. She had calf hypertrophy. At age 5 years, scapular winging, mild proximal lower extremity weakness, and lordosis were evident. Intellectual development and brain MRI were normal. CK levels were >10 times normal. Cardiac evaluation, not described further, was normal. Muscle biopsy revealed dystrophic changes with inflammatory infiltration of macrophages and lymphocytes.
LGMD2O (protein O-linked mannose beta1,2-N-acetylglucosaminyltransferase or POMGNT1).
Two Class III studiese59,e244 and one Class IV studye276 were reviewed. Each study described only one patient, but all studies were retained because of the rarity of the disorder.e59,e244,e276 Age at onset was 12 years in one casee59 and 21 in another.e276 All 3 patients had a limb-girdle pattern of weakness, with neck, hip girdle, and shoulder abductors particularly affected in one.e276 Hamstrings and deltoids were atrophic; calves and quadriceps were hypertrophic.e59,e276 Contractures were absent in one patient and were not described in the others.e59 Cognitive function was normal in all 3 patients. CK levels were elevated >10 times above normal. Cardiac and respiratory involvement were not described. Muscle biopsy in one patient revealed basophilic fibers, some of which were granular, with vacuoles.e276 In another study, the patient with the more severe CMD phenotype was noted to have a severe reduction in the glycosylation of α-dystroglycan compared with the patient with the milder LGMD phenotype.e244
LGMD2P (α-dystroglycan).
One Class IV studye277 reported a 16-year-old Turkish female with LGMD. The clinical features were reported in an earlier study.e278 The patient was born of consanguineous parentage. Age at onset was 3 years with unsteady gait and difficulty climbing stairs. Waddling gait and Gower maneuver were seen at age 10, with microcephaly, increased lumbar lordosis, mild calf hypertrophy, and ankle contractures. Facial weakness or muscle atrophy was not noted. Proximal muscle weakness was observed. Intellectual developmental delay was noted, and IQ at age 16 was 50. CK levels were elevated >10 times at 4,133 U/L and cranial MRI was normal. Muscle biopsy revealed a reduction of α-dystroglycan on immunohistochemistry. The initial description of the patient also reported 8 patients from 7 families with reduced α-dystroglycan expression on muscle biopsy, all characterized by LGMD with onset in the first decade, severe cognitive impairment, and normal brain MRI, but genetic confirmation was not available.e278
LGMD2Q (muscular dystrophy associated with epidermolysis bullosa [plectin-1]).
Thirty articles were reviewed. Most of these were case reports or small case series of up to 4 patients and were included in this guideline because of the rarity of the disorder.e279-e308 This has recently been designated LGMD2Q and is also considered a form of congenital myasthenia.e279,e282,e293,e303,e306 The disorder has been described in several ethnicities, including Dutch, Australian, Japanese, Hispanic, Italian, British, German, and Austrian patients. The characteristic feature was epidermolysis bullosa, in which patients develop blistering of the skin and mucous membranes, usually noted at birth or shortly after. Nail dystrophy was also present. Later in life a progressive muscular dystrophy develops. The onset of dystrophy varied from infancy to as late as the fourth decade, presenting with either hypotonia in the neonatal period, developmental delay, or slowly progressive weakness in late childhood or adulthood. The muscle weakness was described in only a few cases and was in a limb-girdle distribution, with the lower extremity involved more severely.e292,e302 Ptosis and ophthalmoplegia as well as facial weakness were described in a few cases.e283,e292 Dental caries, scarring alopecia, urethral strictures, pyloric atresia, esophageal strictures, respiratory distress, and, rarely, cardiomyopathy were associated features. The frequency of these features was difficult to determine given descriptions of individual cases. CK levels were elevated to varying degrees from <10 times to >10 times normal. Muscle biopsy showed myofiber nuclei in subsarcolemmal rows or clusters. Type I fiber predominance was seen in a few cases. Oxidative stains showed irregular distribution of activity. Myofibrillar disarray and Z-disk streaming were seen on EM. Immunohistochemistry showed loss of sarcolemmal and trace sarcoplasmic activity of the antibody to the rid domain of plectin-1 in type I fibers, whereas type II fibers retained activity. Antibody to the last 50 C-terminal residues of plectin was absent in the sarcoplasm and showed only slight immunoreactivity in both type I and type II fibers in one study.e293
BMD.
We reviewed 3 Class I studiese309-e311 and 54 Class III studies.e65,e66,e68,e156,e208,e312-e360 Bushby et al.e311 is reviewed along with Bushby and Gardner-Medwine309 and Bushby et al.e310 because the former is an earlier report of the same cohort. In a 2-part Class I natural history study of 67 patients with BMD,e309,e310 the age at onset ranged from 10 months to 38 years, with a mean of 11.2 years. Four patients (6%) with genetic confirmation were asymptomatic. Six of 67 (18%) used a wheelchair (mean age of wheelchair dependency was 37.6 years). Those patients with mutations involving exons 45–47 had prolonged ambulation. Myalgias occurred in 27%, calf pain in 81%, and myoglobinuria in 2%. IQ testing was done in only 6 patients and was noted to be in the low-average range, with verbal performance score discrepancy. Most patients (87.9%) attended a mainstream school, but 6.8% attended a school for those with a learning disability and 3.4% a school for those with a physical disability. ECG was abnormal in 14/34 patients tested. The abnormalities included incomplete right bundle branch block (9 patients), Q waves in V4-6 and aVF (5 patients), left ventricular hypertrophy (4 patients), tall R waves in the right chest leads (3 patients), and nonspecific T-wave abnormalities (3 patients). FVC, measured in 41 patients, was generally reduced compared with the expected change for FVC in a comparable normal population. Serum CK levels ranged from 630 to 35,000 U/L, with a mean of approximately 5,200 U/L (>10 times normal).
The 54 Class III studies are summarized here.e65,e66,e68,e156,e208,e312-e360 Symptom onset occurred from early childhood to late adulthood. The pattern of weakness, when present, was proximalgreater-than-distal, with legs more affected than arms. Some patients manifested with only myalgias or episodic myoglobinuria.e315,e331,e357,e358 Most patients had calf hypertrophy; contractures were late and first appreciated at the ankles. Variability in the severity of the clinical phenotype was seen even within families harboring the same mutation.e330
A mild decrease in IQ on average and learning problems were seen in some patients. In a study of neuropsychological testing of 28 patients, 5 had an IQ less than 70 and 13 had an IQ between 70 and 85. The mean IQ was 87.8 (SD 14.8).e318 Another study reported normal IQ (mean 95.6) in 23 patients. However, in 17 males who were tested, a high prevalence of learning abnormalities was noted (reading problems in 21%, spelling in 32%, arithmetic in 26%).e352 A third study of 28 patients with BMD revealed borderline MR in only one patient and a mean IQ of 85.9.e335 Dilated cardiomyopathy with reduced EF occurred in 4% to more than 70%,
depending on the duration of illness.e66,e156,e312,e315,e316,e319,e321,e322,e324,e328e330,e332,e334,e337,e340,e341,e345,e347,e359,e360
In the largest series (98 patients), more than 40% developed a cardiomyopathy.e315 In addition, nonspecific ECG abnormalities were seen in the majority of patients.e312,e313,e321,e328,e333,e340,e341,e346-e348,e356,e357 There was a wide range of CK levels, from slightly to markedly elevated. Muscle biopsies usually revealed reduced immunostaining for dystrophin, but Western blot was more sensitive and demonstrated reduced size or amount of dystrophin.e66,e315-e317,e320,e321,e331 Abnormal immunostaining to sarcoglycans has also been observed.e343,e347
Females manifesting with dystrophinopathy.
Sixteen Class III studies were reviewed.e65,e328,e356,e357,e361-e372 Hoogerwaard et al.e366 and Hoogerwaard et al.e367 describe the same cohort of patients and are reviewed together. Symptom onset was reported to be between ages 2 and 48 years.e357,e361-e363,e366,e368,e370,e372 Patients manifested most frequently with muscle pains with or without a limb-girdle pattern of weakness; 30%–75% had noticeable calf hypertrophy. Asymmetric weakness was noted in 3/15 patients in one study.e370 In one study, 1 of 5 patients had tight heel cords. Weakness could be severe as in DMD or mild like BMD.e65,e356,e358,e361,e362,e364,e368,e371,e372 In a 10-year study of 197 carriers (152 DMD, 45 BMD), 9 DMD (5.9%) and 3 BMD (6.6%) carriers presented with mild calf pseudohypertrophy and 4 DMD carriers (2.6%) had marked proximal wasting and weakness.e369 Normal cardiac status was observed in 15 (45.5%) of the 33 carriers aged between 5 and 15 years but in only 16 (9.8%) of the 164 carriers older than 15 years (p < 0.001). On the other hand, clinically evident myocardial damage was found in 5 (15.1%) of the 33 carriers aged between 5 and 15 years but in 73 (44.5%) of the 164 carriers older than 15 years (p < 0.001). The authors concluded that cardiac involvement in carriers of DMD/BMD is more frequent with increasing age.e369 In another study, 30 of 264 carriers (11%) to 5 of 15 carriers (33%) of DMD/BMD mutation (not necessarily manifesting carriers) had dilated cardiomyopathy on echocardiography; 50/164 (30.5%) had evidence of hypertrophic cardiomyopathy and 7/186 (4%) had arrhythmia of some type on ECG.e328,e365,e369,e370 CK levels were only slightly to markedly elevated to >10 times normal.e65,e356,e361-e364,e368,e371,e372 Immunohistochemistry on muscle biopsies usually revealed a mosaic pattern for dystrophin.e356,e357,e362,e363,e371,e372 A few studies reported preferential inactivation of the putative X chromosome carrying the normal dystrophin allele in most, but not all, affected females.e362,e363,e372
X-linked Emery-Dreifuss muscular dystrophy/EDMD-X1 (emerin).
Twelve Class III studies were reviewed and are summarized here.e81,e373-e383 There was a wide range in age at onset or detection, varying from 14 months to 62 years across the various studies. European, Japanese, and North American kindreds were described. Gradually progressive weakness and atrophy were most typically described in a humeroperoneal pattern, although pelvifemoral distribution of weakness was also described in some families and scapular winging was not infrequent.e81,e376,e378 Most patients remained ambulatory into late adulthood.e81,e378 Rigidity of the spine with an exaggerated lumbar lordosis and contractures at the elbows and Achilles tendon were characteristic features, but inter-and intrafamilial phenotypic variation was frequent; contractures occurred in some patients in the absence of muscle weakness.e376,e377,e380,e383 Cardiac arrhythmias were an important and prominent clinical feature. Conduction disturbances were common, particularly sinus node dysfunction with varying degrees of atrioventricular block progressing to atrial standstill, or paralysis.e81,e373,e375,e376,e378,e381,e383 Notably, conduction abnormalities did not correlate with the severity of skeletal muscle involvement and were described in several patients without any associated myopathic features, including symptomatic nonsustained ventricular tachycardia in 2 patients.e381 A large percentage of patients among studies required permanent pacemaker implantation: 10/12,e373 3/3,e375 15/23,e81 3/5,e377 4/4,e381 and 7/7,e383 for a combined reported incidence of 42/54 (77.8%). Cardiac conduction abnormalities presented at an early age; pacemaker implantation occurred at a mean age between 20 and 35 years in various studies (range 15–42 years).e81,e373,e375-e377 Female carriers were also found to have a high incidence of cardiac conduction abnormalities, increasing with increasing age: 6/34 (18%) overall, 1/29 below the age of 59 years, increasing to 5/5 over the age of 60 years.e373 Pacemaker requirement was reported in 2/34 (6%),e373 2/9 (22%),e383 and 3/5 (60%)e81 female carriers. Thromboembolic stroke was reported in 4 patients.e81,e383 Sudden cardiac death was reported in 2 studies, including 5/23 subjects (22%) in one long-term follow-up studye373 and 3/33 subjects (9%) in 2 families,e383 occurring at mean ages of 47 yearse373 and 34.7 years,e383 respectively (range 27–67 years).
In contrast to the severe conduction abnormalities, CHF was not a clinical feature of emerinopathy, and echocardiography was often found to be normal.e81,e375,e376 However, abnormalities were not infrequent; characteristically, atrial abnormalities, particularly of the right atrium, predominated.e377,e378,e383 In a 5-year longitudinal study,e377 1/5 patients showed right atrial enlargement (RAE) at the outset whereas 4/5 were normal; over the follow-up period, 2 more patients developed RAE. All 3 patients with RAE progressed to biatrial enlargement with early left ventricular enlargement by 5 years; only one patient had LVEF reduced to 45%. Another study revealed RAE in 7/7 (100%) and biatrial enlargement in 2/7 (29%) but no cardiomyopathy.e383 In one series examining left ventricular function by echocardiography, 6/23 patients (26%) had LVEF <50% and 3 of these had severe LVEF compromise (<35%); 2 were asymptomatic.e378
CK levels ranged from normal to mildly elevated (<3 times normal) and were noted to peak during adolescence and then decline in adulthood. In a study of 33 obligate female carriers, CK levels were normal in 100%.e81,e374-e376,e379 Muscle imaging revealed variable involvement of posterior leg muscles, most severely affecting semimembranosus muscles in the thigh and soleus and medial gastrocnemius in 5/5 patients.e381 Immunohistochemistry on muscle biopsies revealed absent or markedly reduced emerin staining.e81,e377,e379,e382,e383 One study found absence of emerin staining on immunohistochemistry of buccal epithelial cells in 3/3 affected males and reduced amounts of nuclear staining in female carriers.e381 Immunocytochemistry on skin biopsies demonstrated absence of emerin staining, whereas immunoblotting of peripheral blood cells showed absence or reduced intensity of the emerin band.e382 Muscle biopsies in 2 cases from one family showed rimmed vacuoles and tubulofilamentous inclusions typical of inclusion body myopathy.e379
EDMD-X2/scapuloperoneal myopathy (four-and-one-half LIM1 protein or FHL1).
Eleven Class III studies were reviewed.e384-e394 Pedigrees included German, Italian-American, Austrian, Croatian, British, Japanese, and northern European. The age at onset showed a wide range, from infancye389 to the eighth decade,e392 but most commonly occurred between childhood and middle age.e387-e391,e393 The clinical phenotypes included limb-girdle weakness,e387,e388,e390,e393 anterior tibial weakness with early foot drop,e385,e392 scapuloperoneal weakness,e385,e388,e390 and rigid spine syndrome.e384,e386,e390,e394 Prominent biceps atrophy was noted in 2 studies.e391,e393 Athletic hypertrophic appearance was seen in some cohorts, especially early in the course of the disease.e384,e388,e390 Scapular winging was noted to be commone385,e388 but more often was not reported. Dysphagia and dysarthria were rare.e386,e387,e392 Extremity contracturese384,e386-e388 and neck contractures were common.e384,e386,e388,e390,e392,e393 Some patients had a cardiomyopathy.e384,e386,e387,e390 Severe respiratory failure was seen in many patients,e386,e388,e390 particularly early-onset patients.e387
CK levels were normal or elevated to <10 times normal.e384,e387,e388,e390,e392,e393 Muscle biopsies frequently showed reducing bodies and cytoplasmic bodies.e384,e386,e387,e392,e394 Two reports found cytoplasmic bodies without reducing bodies.e384,e388FHL1 immunostaining was used in several studies and noted to be positive for reducing bodies.e384,e387,e390-e392
Myofibrillar myopathies.
The term myofibrillar myopathies (MFMs) is used to describe a group of muscular dystrophies that share specific, common morphologic features on muscle biospy.e395,e396 On light microscopy these abnormalities are best appreciated on the modified Gomori trichrome stains, in which the abnormal fibers harbor an admixture of amorphous, granular, or hyaline deposits that vary in shape and size and are dark blue or blue red in color. Many abnormal fiber regions, especially the hyaline structures, are devoid of or have diminished oxidative enzyme activity. Some hyaline structures are intensely congophilic. Some muscle fibers harbor small rimmed vacuoles. EM shows that disintegration of the myofibrils begins at the Z-disk, followed by accumulation of degraded filamentous material in various patterns, aggregation of membranous organelles and glycogen in spaces vacated by myofibrils, and degradation of dislocated membranous organelles in autophagic vacuoles. Immunostaining reveals ectopic accumulation of multiple proteins, including myotilin, αB-crystallin, desmin, dystrophin, sarcoglycans, caveolin, neural cell adhesion molecule, plectin, gelsolin, ubiquitin, filamin C, Bag3, and others. One study reported differences between the distinct MFM subgroups: the consistent presence of “rubbed-out” fibers in desminopathies and αBcrystallinopathies, an elevated frequency of vacuoles in ZASPopathies and myotilinopathies, and the presence of a few necrotic fibers in myotilinopathy patients.e397
In a separate study, the same group of authors reported that EM findings in desminopathies and αB-crystallinopathies were very similar and consisted of electron-dense granulofilamentous accumulations and sandwich formations; they differed in the obvious presence of early apoptotic nuclear changes in αB-crystallinopathies.e398 ZASPopathies were characterized by filamentous bundles (labeled with the myotilin antibody on immune-EM) and floccular accumulations of thin filamentous material. Tubulofilamentous inclusions in sarcoplasm and myonuclei in combination with filamentous bundles were characteristic for myotilinopathies. Rather than reiterating the hallmarks above in each of the subsequent MFM subtypes, we note that each of these disorders shows the characteristic MFM features on biopsy.
Myotilin (LGMD1A).
Nine Class III studies were reviewed.e9,e399-e406 The age at onset was 18–79 years with limb-girdle or distal limb weakness. Asymmetric muscle weakness and atrophy were frequently noted across studies. Scapular winging was not reported. Dysarthria was found in 4/16 affected individuals of a single kinship who were assigned as having LGMD1Ae402 and in the most-affected family members of another large kinshipe401; another patient had hypernasal speech.e403 Ten of 16 in the initial reporte407 (cross-referenced in Hauser et al.e402), 1/5,e9 and 4/13e403 patients had tight heel cords. Cardiac abnormalities were seen in 3/5,e9 2/13,e403 1/24,e406 and 1/12e400 patients. Respiratory failure was present in 3/13,e403 3/24,e406 and several affected family members of a large kinship.e401 Serum CK levels were normal to <10-fold elevated. Muscle-imaging studies revealed involvement of the medial gastrocnemius, soleus, hip adductors, and biceps femoris with fatty/fibrous replacement and edema; the semitendinosus was relatively spared.e399,e400,e403,e405,e406 Muscle biopsy showed features of MFM. Myofiber necrosis and inflammatory infiltrates were also seen occasionally.e9,e399,e403,e406
Desmin (LGMD1E).
There were 17 Class III studies.e400,e405,e406,e408-e421 The myopathy was usually inherited in an autosomal dominant fashion, although sporadic cases were reported. Age at onset ranged from the first to the sixth decade of life. The pattern of muscle weakness was most often distal-greater-than-proximale409,e411-e413,e415,e417,e418 with the earliest manifestation being progressive foot drop, but proximal-greater-than-distal weakness,e413,e420 proximal and distal weakness,e408-e410,e413,e415-e417 and a scapuloperoneal distributione413 were also reported. Face and bulbar muscles were affected in some with dysphagia or dysarthria.e408,e412,e413,e415-e418,e420 Ventilatory muscle weakness was also common.e411,e413,e415,e417,e418 Cardiac involvement was common (40%–100%) across all series, with onset often preceding muscle weakness.e400,e406,e408-e410,e412-e418,e420
Onset of cardiac symptoms ranged from the first to the seventh decade of life. The most common cardiac manifestation was cardiac arrhythmia, including atrioventricular conduction block, atrial fibrillation, other tachyarrhythmias, and cardiac conduction defects, and some patients required pacemaker or implantable cardioverter defibrillator implantation. Dilated cardiomyopathy was more frequent than hypertrophic or restrictive cardiomyopathy. In a study of 21 members of a Swedish family, arrhythmogenic right ventricular cardiomyopathy was described in 3, and the authors suggested that the presence of a right ventricular cardiomyopathy (right ventricular dysfunction and tachyarrhythmias of right ventricular origin) was a clue to this disorder.e421 Sudden death was reported in 3/18 patients, and 2/18 patients underwent cardiac transplantation in one series.e406
Serum CK levels were normal or only moderately elevated (up to 5 times normal) in the majority of patients.e409-e412,e416,e418 Two studies focused on CT/MRI of skeletal muscles.e400,e405 One study evaluated 19 patients and revealed gluteus maximus greater than gluteus medius or gluteus minimus involvement.e400 In the thighs, the semitendinosus, sartorius, and gracilis were affected earlier than the adductor magnus, biceps femoris, or semimembranosus. The quadriceps muscles were relatively spared. In the distal legs, the peroneal muscles were affected more than the tibialis anterior, which was affected more than the posterior compartment. The other study evaluated 4 patientse410 and reported that the iliopsoas, sartorius, gracilis, and semitendinosus were affected in 3 of 4 patients, whereas the biceps femoris was involved in 2 patients and the semimembranosus in one. In the distal legs, the peroneal, tibialis anterior, medial gastrocnemius, and soleus were involved in 3 of 4 patients. Two patients had involvement of the paraspinal muscles and 2 of the shoulder girdle, whereas none had involvement of the humeral muscles in the arms. Another CT study of 4 patients showed the earliest abnormalities in the semitendinosus and sartorius, and later abnormalities in the gracilis muscles at the mid-thigh level and in the peroneal group followed by the anterior tibialis and posterior group at the mid-calf level.e417 In the later stages of the illness all muscles except for the soleus were replaced by fatty tissue.
αB-Crystallin.
Four Class III studies presenting a total of 9 patients were reviewed.e400,e422-e424 The age at onset ranged from the 30s to the late 60s. Two patients presented with distal lower limb weakness with asymmetric muscle atrophy; one patient presented with diaphragmatic weakness followed by leg weakness and dysphagia.e422,e423 Five patients from the same family had dysphagia and dysphonia, and 4 of them had cataracts.e424 Scapular winging, dysarthria, or contractures were not described. Three patients had cardiomyopathy. Serum CK levels were normal to <10-fold elevated. Muscle-imaging studies showed involvement of gluteus maximus, sartorius, semitendinosus, vastus intermedius, medialis, lateralis, rectus femoris, tensor fasciae latae, adductor magnus, gracilis, and peroneal muscles.
Z-band alternatively spliced PDZ motif-containing protein (ZASP) (Markesbery-Griggs distal myopathy).
Four Class III studies were reviewed.e72,e400,e406,e425 The age at onset was 27 to 73 years. Patients presented with limb-girdle or distal lower limb weakness. In one study, all 10 patients from a single family had distal leg weakness at onset, followed by atrophy and weakness of the hand muscles and wrist extensors.e425 In another study of 3 patients, one had distal weakness and 2 had distal-greater-than-proximal weakness.e400 Likewise, a third study had 5/11 patients with distal-greater-than-proximal weakness; only distal weakness occurred in one patient, only proximal weakness in 2 patients, and both proximal and distal weakness in 3 patients.e72 One report of 7 patientse406 also revealed distal-predominant weakness at onset that spread to involve distal and proximal muscles as the disease progressed. Scapular winging, dysphagia, or dysarthria was not noted. Ankle contracture was observed in one of 10 patients (10%) in one study.e425 Cardiomyopathy was reported in 3 of 11 patients.e72 Serum CK levels were normal to <10-fold elevated. Muscle MRI showed early involvement of posterior calf muscles, and the soleus muscle was the most affected. In the pelvis, the gluteus minimus was most affected in 3/3 patients. At the thigh level, the posterior compartment (biceps femoris and semimembranosus) was mostly involved, whereas the adductor magnus and gracilis were relatively spared. In the lower legs, one patient presented only with alterations in the soleus and medial gastrocnemius muscle at disease onset; in another case the soleus was most affected, and in the third case all lower leg muscles were involved.e400 Muscle MRI performed in 2 patients in another study showed considerable involvement of posterior calf muscles.e425 In another study, the adductor magnus, semimembranosus, vastus medialis, biceps femoris, soleus, and gastrocnemius were most frequently involved.e406 Five of 11 patients had clinical or biopsy features of an associated neuropathy.e72
BCL2-associated athanogene 3 (BAG3).
Two Class III studies describing 7 patients were reviewed.e73,e426 One studye73 reported 2 patients (one age 15 years and one age 11 years) presenting with a history of toe walking, the former since early childhood and the latter since he was a toddler. The third patient presented at age 13 with scoliosis, rigid spine, and easy fatigability. The distribution of weakness was axial and moderately severe, with distal-greaterthan-proximal weakness in one patient, moderate proximal weakness in one patient, and severe diffuse weakness in the third patient. One patient had scapular winging. Another patient had hypernasal speech. One patient had knee and ankle contractures. Two patients had restrictive cardiomyopathy, and one had mitral regurgitation. All 3 patients had respiratory involvement. CK levels were 3- to 15-fold elevated. Another studye426 reported 4 patients from 3 families with weakness starting at ages 5 to 12 years. Two had predominantly proximal muscle weakness, restrictive/hypertrophic cardiomyopathy, and respiratory insufficiency; one developed bilateral pes cavus, neck and sital leg weakness, and restrictive cardiomyopathy with secondary enlargement of both atria, and the other developed restrictive cardiomyopathy, had a heart transplant at age 13 years, and became ventilator dependent. At age 15, he was noted to have predominantly proximal and respiratory muscle weakness.
Filamin C (Williams distal myopathy).
Seven Class III studies were reviewed.e400,e427-e432 Fischer et al.e400 described muscle MRI findings of some of the patients previously reported in Kley et al.,e428 so these 2 studies are reviewed together. Mean age at onset of weakness was early- to mid-40s. The patients presented with a limb-girdle pattern of proximal muscle weakness and atrophy with lower limb predominance; 6 of 31 patients had scapular winging. No dysphagia, dysarthria, hoarse voice, or contractures were noted. In a study of 13 patients from 3 related families, all patients presented with distal upper limb weakness with lower limb involvement upon disease progression.e431 There was late-onset involvement of respiratory muscles in 14/31 patients, but no details were provided. Cardiac involvement was seen in approximately one-third, including atrial flutter, right bundle branch block, decreased EF, and nonspecific cardiomyopathy. CK elevations were mild (<10-fold). Muscle MRI showed significant sparing of sartorius, gracilis, superficial parts of the quadriceps femoris, and the lateral gastrocnemius. There was involvement of gluteal muscles, semimembranosus, adductor magnus, biceps femoris, and vastus intermedius and medialis. The soleus and medial gastrocnemius were disproportionately involved compared with the lateral gastrocnemius and peroneal muscles. In one study, MRI of a single patient showed marked triceps surae with involvement of other lower leg muscles, with the exception of the posterior tibial muscle.e431
In a study of a Chinese family with 10 affected members over 4 generations, proximal muscle weakness and atrophy of the lower limbs were noted at the onset; as the disease progressed, all limbs were involved.e432 The index patient had right bundle branch block and atrial and ventricular premature beats.
In a Class III study originally categorized as the Williams distal myopathy and subsequently confirmed to be due to filamin C mutations,e429,e430 12 affected members of a single Australian kindred were described with onset of distal upper and lower extremity weakness in early adulthood. Two additional family members were possibly affected. Age at onset varied considerably, but all cases reported onset by the fourth decade of life, and in many cases much younger: 5/27 reported onset in their teens and 4/27 reported onset around age 30 years. The characteristic pattern included early involvement of the distal anterior upper limb (selectively involving forearm pronators and finger flexors) and posterior leg (ankle plantar flexor) muscles, with sparing of the tibialis anterior, even in advanced disease. Muscle pain was a prominent feature. All patients remained ambulatory. None had evidence of cardiac or respiratory muscle involvement. Serum CK levels were either normal (5/8) or mildly elevated (<3 times normal in 3/8 patients). MRI showed widespread involvement of the posterior and lateral leg compartments in 4/7 patients (57%) and was normal in 3/7 patients (43%). In a similarly affected Italian family, the weakness started distally in the hand muscles with hand muscle atrophy and slowly progressed to involve the proximal muscles. Two of the 3 patients had cardiomyopathy.e430
Muscle biopsies showed features of MFM except for patients in Duff et al.e430 and Guergueltcheva et al.,e431 whose biopsies showed nonspecific myopathic changes.
Titin.
This is described under the section on LGMD2J/Udd distal myopathy/HMERF.
Hereditary inclusion body myopathies.
Autosomal recessive hIBM/Nonaka distal myopathy (GNE). Eleven Class III studies were reviewed.e433-e443 In addition, another report (Sadeh et al.e444) before confirmation by genetic testing was also reviewed in conjunction with MitraniRosenbaum et al.e434 Autosomal recessive hIBM (AR-hIBM) and Nonaka distal myopathy are the same disorder caused by mutations in the gene encoding UDP-N-acetylglucosamine-2epimerase/N-acetylmannosamine kinase (GNE). AR-hIBM was initially described in Iranian Jews and other Middle Eastern Karaites and Arab Muslims of Palestinian and Bedouin origin, whereas Nonaka distal myopathy was reported in Japanese, Korean, and Chinese families.e436-e439 The age at onset ranged between the late teens and early 40s.e434,e436,e441 The characteristic pattern of muscle involvement was early involvement of the anterior tibial muscles leading to progressive foot drop. Over time, proximal legs could be involved, but there was relative sparing of the quadriceps in comparison to sporadic inclusion body myositis. The extensor muscles in the forearms also become affected, followed by involvement of more proximal arm muscles.e434,e436,e441 Mild neck flexor weakness was noted in some patients (number not mentioned), and facial weakness was noted in 2/55 families in one study.e436 Bulbar and extraocular muscles were spared.e434 The heart was not typically involved, but dilated cardiomyopathy developed in 2 patients late in their course.e433 Serum CK levels were normal or mildly to moderately elevated (2- to 6-fold).e436,e441 Muscle ultrasound of 6 patients demonstrated severe atrophy of the hamstring, anterior tibial, and peroneal muscles with milder quadriceps and gastrocnemius involvement and central atrophy with peripheral sparing; the “myopathic target” was noted in all 6 patients in the hamstrings.e435 Muscle biopsy demonstrated dystrophic myopathy and rimmed vacuoles. The autophagic vacuoles had nuclear and cytoplasmic 15- to 18-nm filamentous inclusions on EM.e437-e439,e441-e443
Perivascular lymphocytic inflammation was described in 1/55 families in one study.e436
Autosomal dominant hIBM with Paget disease and frontotemporal dementia (hIBMPFD) (valosin-containing protein or VCP).
Seventeen Class III studies were reviewed and are summarized here.e445-e461 Pedigrees included Asian, North American, European, Scottish, British, and Australian families. The myopathy was variably associated with Paget disease of bone (PDB), frontotemporal dementia, and more recently motor neuron disease (familial amyotrophic lateral sclerosis [fALS]). There was significant heterogeneity in clinical phenotype and severity both between and within families. Age at onset was variable, but across studies the mean age at onset for myopathy and PDB was 43 years (range late 20s–81 years). Most patients presented with either limb-girdle or scapuloperoneal weakness. A purely distal myopathy affecting lower and upper extremities was reported a Finnish kindred.e461 Scapular winging and lumbar lordosis were common. Frontotemporal dementia was described in approximately 30%–50% of patients, with onset approximately 10 years after weakness (average age 54 years). PDB tended to occur earlier than in sporadic PDB and was seen with variable frequency, ranging from 29% to 100% in various kindreds; in some patients, PDB was not clinically apparent but was diagnosed by laboratory and radiographic findings. In a British pedigree, dilated cardiomyopathy (4/18 patients), urge incontinence (5/5 patients), and fecal incontinence (4/5 patients) were described.e451
CK levels were normal or slightly elevated (<10-fold). Elevated blood alkaline phosphatase was found with high frequency among patients with PDB (86% average across the studies [range 57%–100%]). Muscle MRI in 2 studiese451,e454 showed symmetric fatty degeneration of the quadriceps/hamstrings/glutei and anterior/posterior compartment of the legs. In the upper extremity, MRI showed fatty degeneration of paraspinal, supraspinatus, infraspinatus, and teres minor, and less so biceps, triceps, and deltoid.
Characteristic findings on muscle biopsy included rimmed vacuoles with ubiquitin and VCPpositive cytoplasmic inclusions, although in the larger series these were noted in less than half of biopsies (33%–39%).e447,e448,e460 Most biopsies revealed nonspecific myopathic abnormalities as well as neurogenic features of type grouping and angulated fibers. The latter may be due to fALS that can be associated with VCP mutations.e447,e448,e460 EM showed paired helical filaments in muscle and in PDB osteoclasts.e460
Fast myosin heavy chain, MYHC-IIA, IBM3.
Five Class III studies were reviewed.e462-e466 Since some of the Swedish studiese463,e465 appeared to report the same patients, they were reviewed along with Martinsson et al.,e466 the original study. This disorder has been reported in Sweden, Finland, and the United Kingdom.e463,e464,e466 Age at onset ranged from birth to 40 years.e463,e464 Myalgia and muscle weakness were the presenting symptoms in 7/15 patients in one familye463 and were common in the first report of 19 patients as well, although numbers were not provided.e466 Muscle weakness was predominantly proximal in a limb-girdle distribution. External ophthalmoplegia was a consistent finding in all 19 patients of one family in the initial report. They were found to have a dominant missense mutation, p.E706K,e466 but ophthalmoplegia was not described in a second report of 8 patients who had different missense mutations in the MYHC-IIA gene. In patients in whom the disease started at birth, congenital joint contractures were seen.e465,e466 Hand and face weakness were noted in 7/19 patients, and congenital hip dislocation in 4/19.e466 Mild cervicothoracic kyphoscoliosis was described in 3/15 patients in one studye463 and 7/19 in the original description of one family.e466 Finally, one study described 5 patients with nonsense or truncating mutations from 3 families, one from the United Kingdom and 2 from Finland.e464 Three presented in early childhood; 2 were asymptomatic. All had pronounced ophthalmoplegia, ptosis, and facial muscle weakness. Neck flexors were weak in 3/5, elbow flexors and ankle dorsiflexors were weak in 2, and 3 had joint hypermobility. Serum CK levels were normal in all 3 patients in whom they were checked. Two patients had muscle MRI that showed moderate diffuse fatty degenerative changes in the thigh and medial gastrocnemius. Muscle biopsy in the initial description revealed small and infrequent type II fibers, focal disorganization of the myofibrils, and rimmed vacuoles and inclusion consisting of 15- to 20-nm tubulofilaments.e466 One muscle biopsy had lobulated fibers and another had central nuclei and minicores in another study of patients with a different missense mutation.e463 Finally, in patients with the nonsense or truncating mutation, muscle biopsies demonstrated the absence of type II A fibers and myopathic changes.e464 Thus, different mutations in the MYH-IIA gene appear to have different clinical presentations and muscle biopsy features.
Distal muscular dystrophies/myopathies.
Welander distal myopathy. Six Class III studies were reviewed.e263,e467-e471 Although most often seen in the Swedish population,e468 Finnish patients with Welander distal myopathy have also been described.e471 This myopathy was recently reported to be caused by mutations in the RNA-binding protein described initially as T-cell restricted intracellular antigen (TIA1), now known to be expressed widely.e472 The age at onset ranged from 24–60 years across studies, with a mean in the third to fourth decade of life.e263,e468e471 The weakness predominantly involved the finger extensors and foot dorsiflexors and often began in either the hands or the legs. All 7 patients had foot dorsiflexor weakness in one study.e467 All 9 patients in another study had finger extensor weakness, and 7/9 patients had foot dorsiflexor weakness.e468 In a third study, 2/4 patients had finger extensor weakness, 1/4 had both finger extensor and foot dorsiflexor weakness, and the remaining 1/4 had weakness limited to foot dorsiflexors.e470 Thenar muscle atrophy was noted in 3/4 patients in one study.e470 An unusual feature of Welander myopathy is impaired distal sensatione468,e469,e471; however, nerve conduction studies were normal in all patients in one study.e469
CK levels were normal in 3/4 patients and mildly elevated (<10-fold) in 1/4 patients.e263,e470 Imaging studies (CT and MRI) revealed fatty infiltration in the tibialis anterior, gastrocnemius, and soleus muscles with relative sparing of the peroneus and tibialis posterior muscles.e263,e467,e471 In the proximal leg, the biceps femoris, semitendinosus, semimembranosus, and adductors were involved. Muscle biopsy revealed rimmed vacuoles mainly in atrophic fibers but also less frequently in normal fibers when performed in the distal muscles such as the tibialis anterior.e468e471 In contrast, biopsy of a proximal muscle such as the vastus lateralis was normal in 7/7 patients in one study.e467 EM revealed cytoplasmic 16- to 21-nm filaments associated with the rimmed vacuoles. Other abnormalities included dense collections of Z-disk material or streaming, double Z-disks, honeycomb material, and abnormal mitochondria.e470 Moderate loss of myelinated nerve fibers was noted in 2/5 patients in one study.e469
Markesbery-Griggs distal muscular dystrophy.
This is discussed in the MFM section under ZASP.
Udd distal muscular dystrophy.
This is discussed in the section on LGMD2J and HMERF (titinopathy).
Miyoshi distal myopathy.
This is discussed in the sections on LGMD2B (dysferlin) and LGMD2L (anoctamin-5).
Nonaka distal myopathy.
This is discussed in the section on AR-hIBM/GNE.
Laing myopathy/MYH7.
Eleven Class III studies were reviewed.e473-e483 Mastaglia et al.e475 is a follow-up of the kindred described in Laing et al.e478; these are reviewed together. Disorders caused by myosin heavy chain (MYH7) mutations have been classified into 2 subgroups with distinct clinical and pathologic findings: Laing distal myopathy (LDM) and hyaline body (or myosin storage) myopathy. In LDM, the typical clinical features are of an early-onset distal myopathy, with weakness and atrophy beginning in the first decade and selectively involving the anterior compartment of the lower leg, including ankle and toe dorsiflexors, resulting in foot drop or “hanging big toe.” Weakness progresses slowly, with an average of 10 years before involvement of finger extensors and neck flexors, and later development of proximal extremity weakness.e474,e475,e478,e483 Neck flexor weakness is a distinguishing feature from other distal myopathies and was reported in 20%–100% of cases in various series.e474,e478,e483 Mild facial weakness was described in up to 70% of patients.e475,e477 Calf hypertrophy was noted in 11/31 (35%) in the largest cohort of patients.e477 Scapular winging was observed in 2/9 (22%) and 4/31
(13%) patients in the largest cohorts.e477,e478 Disabling myalgias were a clinical feature in 33% of 31 patients.e477 Associated findings of scoliosis, pes cavus, ankle contractures, and lumbar hyperlordosis were variably described in about half of 43 patients reported.e474,e476,e477,e483 One of 27 patients had dilated cardiomyopathy,e477 but this was atypical; a single patient with a syndrome of hypertrophic cardiomyopathy and tibialis anterior hypertrophy was described.e480 Skeletal muscle CT scans in 7 patientse477and MRI in 28 patients revealed selective early involvement of the toe extensors, anterior tibialis, and sternocleidomastoids.e474,e477,e478,e483 Muscles from the posterolateral lower leg and rectus femoris were affected at very late stages; the gastrocnemius was spared.e477Across studies, serum CK levels were normal or only slightly elevated (<3 times the ULN).e474,e477,e478,e480 Five studies described muscle biopsies in 21 patientse474,e477,e478,e480,e483; these showed variable and often nonspecific myopathic changes, most commonly atrophy and type I fiber grouping. Rimmed vacuoles were typically absent. Myosin immunohistochemistry demonstrated coexpression of slow and fast myosin in type I fibers, highly characteristic of LDM.
Four studies were relevant to myosin storage (hyaline body) myopathy.e473,e479,e481,e482 Although phenotypes vary considerably, muscle biopsy findings are characteristic for the disorder. Patients presented from the first to the fifth decade, and the average age at onset ranged from 29–38 years.e479,e481 Weakness in either a scapuloperoneal or a limb-girdle distribution was described; CK levels were normal or slightly elevated.e473,e479,e481,e482 In a British kindred, limb-girdle weakness was associated with hypertrophic cardiomyopathy in 3/3 siblings,e481 but other patients were reported to have normal echocardiography.e473,e482 Muscle biopsies from 8/8 individuals revealed subsarcolemmal “hyaline bodies”: discrete, amorphous, eosinophilic material in type I fibers exclusively, staining homogeneous pale pink on hematoxylin & eosin and faint green on Gomori, which stains intensely with antibodies to MHY7.e473,e479,e481,e482
Vocal cord and pharyngeal weakness with distal myopathy (matrin-3).
Two Class III studies were reviewed.e484,e485 Two pedigrees were reported: one white North American and one Bulgarian. Mean age at onset of weakness was 45 years. The clinical phenotype was slowly progressive, beginning with foot drop and distal upper extremity muscle weakness and progressing to involve proximal muscles, frequently associated with dysphagia and dysphonia. One patient from the North American pedigree (37 individuals, 12 who underwent clinical examination) had restrictive ventilatory weakness and low EF. None of the patients in this pedigree had unexplained cardiomyopathy. CK levels were slightly elevated (<10 times normal) in 13/17 cases tested and normal in 4/17. Muscle biopsies showed characteristic subsarcolemmal rimmed vacuoles in 5/7; 2/7 biopsies showed end-stage changes.
Filamin C (Williams distal myopathy).
This is discussed in the MFM section on filamin C (Williams distal myopathy).
Nebulin (NEB).
Two Class III studies were reviewed.e486,e487 The first Class III study described 7 patients of Finnish descent from 4 unrelated families with a novel recessively inherited distal myopathy caused by homozygous missense mutations in the nebulin gene.e486 In the second Class III study, 3 non-Finnish patients from 2 unrelated families were reported to have distal myopathy caused by 4 different compound heterozygous nebulin mutations. Onset was in early childhood, with very slowly progressive weakness and atrophy of ankle dorsiflexors, finger extensors, and neck flexors, initially presenting with foot drop. Delayed motor milestones (walking at 2 years), an elongated face, and lumbar lordosis were reported in 2 children who presented at ages 11 and 13 years with foot drop.e487 Mild to moderate facial weakness was noted in 6 of 7 patients (86%).
CK levels were normal in 6/7 patients (86%) and slightly elevated in 1/7 patients (14%) in the Finnish study and normal in all 3 patients of non-Finnish descent.e486,e487 One of 6 patients (17%) demonstrated a low FVC.e486 Imaging was abnormal in all patients: CT (3/3) and MRI (5/5) of the leg muscles showed fatty degeneration in the anterior compartment of the lower legs.e486,e487 Muscle biopsies demonstrated nemaline bodies.e486,e487
Distal myopathy with Kelch-like homologue 9 mutations (KLHL9).
One Class III studye488 reported a German kindred with 10 affected members who had weakness and atrophy of the anterior tibial muscles (age at onset between 8 and 16 years), followed later by atrophy of the intrinsic hand muscles. The disorder was slowly progressive and patients retained the ability to walk until the seventh decade. Ankle contractures were present in all 10 patients. Tendon reflexes were absent in the lower extremities in 2 patients. Reduced sensation in a distal symmetric pattern was noted in 7 patients between the ages of 25 and 67 but not in the younger patients.
CK levels were normal or mildly elevated in 8 patients and moderately elevated (144 U/L) in one. Nerve conduction studies in 2 patients revealed mildly prolonged distal latencies in some motor nerves and reduced sural amplitude in one patient. ECG, echocardiogram, and pulmonary function tests were normal in one patient. MRI of the lower extremity in the index patient showed symmetric fatty atrophy that was most prominent in the semimembranosus, biceps femoris, and vastus intermedius, whereas the vastus lateralis and medialis, sartorius, gracilis, and adductors were preserved. The tibialis anterior, gastrocnemius, and soleus were more affected than the peroneus longus or tibialis posterior. Muscle biopsy in the index case did not reveal vacuoles. There were dystrophic changes and angulated fibers. There was loss of fiber typing on NADH stain. Sural nerve biopsy did not reveal neuropathy. Immunohistochemistry revealed normal expression of dystrophin, caveolin-3, laminin-α2, and sarcoglycan-dystroglycan complex. All patients possessed a heterozygous mutation in the KLHL9 gene encoding a bric-abrac Kelch protein.
Other disorders:
Selenoprotein (SEPN1, rigid spine syndrome).
Most studies reviewed discussed the congenital myopathy phenotype of SEPN1 mutations and are not included in this guideline. Two Class III studies are reviewed.e85,e489 One large Class III studye489 described the clinical course of SEPN1-related muscular dystrophy in 41 patients. Mean age at onset was 2.7 years and ranged from birth to the second decade. In 19 of 41 patients (46%), the onset was between 6 months and 5 years, with delayed milestones, difficulty running, or falls. Scoliosis or easy fatigability was noted at onset in 3 children between the ages of 6 and 10 years; 2 presented at 7 years with running difficulty and 2 at age 13 years with back stiffness and generalized muscle weakness and atrophy. CK levels were normal in 29/37, minimally elevated in 6/37, and markedly elevated in only 1/37. Rigid spine was noted in 25 patients and scoliosis in 26 with onset between the ages of 1 and 20 years. Nine patients required spinal surgery by age 15 years, and an additional 4 required it by age 20. Joint contractures were present in 26 patients by the age of 10 years and involved the Achilles tendon, elbow, and long finger flexors. Mild right ventricular hypertrophy/pulmonary hypertension was found in only 5 patients. Twenty-seven of 41 patients required nocturnal noninvasive ventilation for reduced FVC. Most patients remained ambulatory; only 6 became wheelchair dependent. MRI in 13 children revealed selective or prominent involvement of the sartorius; no selectivity was noted at the calf level.e85 Muscle biopsy features included nonspecific myopathic change (5/20), type I fiber predominance (5/20), multiminicores (8/20), and cores (2/20); Mallory bodies were noted in one patient who also had nonspecific myopathy.
Muscular dystrophy with generalized lipodystrophy (cavin-1/polymerase I and transcript release factor [PTRF].
Three Class III studies were reviewed.e490-e492 The first study reported 5 Japanese patients of nonconsanguineous parentage with PTRF mutations causing a secondary deficiency of caveolin-3.e490 The second studye491 reported 15 patients from Oman and one from the United Kingdom. The third study reported 2 Mexican siblings and a Turkish girl with the disorder.e492 The age range was from the neonatal period to 24 years. All patients had generalized lipodystrophy. Clinical information was incomplete in the second study.e491 Developmental delay was noted in all 3 patients in one study.e492 Weakness was present in 12/15 patients in the second studye491 but not described. In the first study, weakness was distal dominant in 2/5, generalized in one, and absent in the other 2.e490 Almost all patients had electrically silent percussion-induced muscle mounding, muscle hypertrophy, myalgias, and cramps. Five of the 23 described patients had cardiac arrhythmias, including prolonged QTc syndrome. Sudden death in the teenage years occurred in 4 patients in one study.e491 Six of 23 patients had hepatosplenomegaly/fatty liver.
Across studies, scoliosis was noted in 5 patients, rigid spine in 2, lordosis in one, and contractures (ankle, finger, and other unspecified) in 6. Fifteen patients had congenital hypertrophic pyloric stenosis. Insulin resistance (4 patients across the studies), elevated serum triglycerides (10), acanthosis nigricans (3), and atlantoaxial dislocation (1) were other features. CK levels were elevated in all patients tested, ranging from 542–2,630 IU/L (<10- to >10-fold). Muscle CT scan in one patient revealed hypertrophy of paravertebral and thigh muscles with minimal subcutaneous and abdominal fat.e490 Reduced immunoreactivity to PTRF antibodies was noted on muscle biopsies. Caveolin-3 immunoreactivity was greatly reduced in the sarcolemma, but cytoplasmic staining was increased in a pattern similar to LGMD caused by caveolin-3 mutations. Immunoblotting revealed absent PTRF bands.e490
Conclusion
In patients with LGMD and distal muscular dystrophy, a combination of clinical, radiologic, and laboratory features are useful in directing genetic diagnosis (multiple Class I–III studies). Single features that are pathgnomonic of a disorder are seen only rarely (see figures 1–5 in the summary article published in print and figures e-1 and e-2 and table e-2, available as online data supplements).
Clinical Question 5: Are there effective therapies (medications, gene therapy, exercise, complementary and alternative therapies, orthopedic interventions, surgery) for muscular dystrophies that improve muscle strength, slow the rate of strength decline, preserve ambulation and overall function, delay time to tracheostomy ventilation, maintain healthy EF, slow cardiac mortality, preserve quality of life and activities of daily living, and delay overall mortality?
There were 12 studies (2 Class I,e493,e494 4 Class II,e495-e498 and 6 Class IIIe246,e344,e462,e465,e499,e500) evaluating treatments for disorders described below. No articles were identified for the other disorders discussed in this guideline.
In a Class II randomized, double-blind, placebo-controlled trial,e498 adeno-associated virus (AAV) gene transfer to the extensor digitorum brevis (EDB) muscle was performed in 6 patients with LGMD2D (α-sarcoglycanopathy) on one side. Saline was injected into the opposite EDB as a control. α-Sarcoglycan gene expression increased 4- to 5-fold in 3 subjects (at 6 weeks in 2 subjects and at 12 weeks in the third subject); restoration of the α-sarcoglycan complex was also noted.e498 Three subjects were followed for an additional 3 months and reported on in a subsequent Class I study.e494 Persistent α-sarcoglycan gene expression was noted at 6 months in 2/3 subjects. One patient failing gene transfer demonstrated an early rise in neutralizing antibody titers and T-cell immunity to AAV. No adverse effects (AEs) were reported.e494
A Class III studye500 of 3 patients with LGMD2C who received the highest of 3 escalating doses of AAV-vector expressed human γ-sarcoglycan genes (4.5 x 1010 copies) into the extensor digitorum communis found increased γ-sarcoglycan expression (4.5%–10% positively stained fibers on muscle biopsy) 30 days after injection. One patient had detected γ-sarcoglycan by Western blot as well. Muscle strength was stable over 6 months of follow-up. MRI of the forearm before and 15 days postinjection did not reveal changes. Inflammatory markers did not change and no inflammation was noted on repeat biopsy.
Conclusion.
AAV gene therapy into the EDB muscle of patients with LGMD2D probably increases the expression of α-sarcoglycan gene and restores the protein complex for up to 6 months post-injection without significant AEs (one Class I and one Class II study). Data are insufficient to determine the effect of AAV-vector expressed γ-sarcoglycan genes in patients with LGMD2C (one Class III study).
Clinical context.
These are small proof-of-concept studies. Despite increased expression of the target protein in these few patients, the clinical relevance of gene therapy is yet to be determined. Other considerations include the number of injection sites and frequency of injection.
A Class I phase 1/2 randomized, double-blind, placebo-controlled trial evaluated the safety and tolerability of a neutralizing antibody to myostatin (MYO-029), which is an endogenous inhibitor of muscle growth.e493 Four dosing cohorts of 36 patients each (total 116 subjects: 36 BMD; 38 LGMD2A, 2B, 2C, 2D, 2E, and 2I; 42 facioscapulohumeral dystrophy) were included in the trial. The dosing cohorts were 1 mg/kg, 3 mg/kg, 10 mg/kg, and 30 mg/kg intravenously every 2 weeks for 6 months, for a total of 13 doses. Subjects were followed for 3 months after the last dose. MYO-029 was found to be safe and well tolerated. One hundred four of 116 subjects (89%) reported AEs. The only AE that was significantly more common in the treatment group was accidental injury (8/27 [27.6%] in the placebo group and 13/27 [48%], 11/27 [41%], and 4/27 [15%] in the 1, 3, and 10 mg/kg cohorts, respectively, p = 0.026). The major AE was cutaneous hypersensitivity, seen in 4/27 patients (15%) in the 10 mg/kg group and 2/6 patients (33%) in the 30 mg/kg cohort. Rash and urticaria were noted in 12 subjects in all (2/29 [7%] in the placebo group, 3/27 [11%] in the 1 mg/kg group, 1/27 [3.7%] in the 3 mg/kg group, 4/27 [15%] in the 10 mg/kg group, and 2/6 [33%] in the 30 mg/kg group). Seven subjects (6%) had serious AEs (2/29 patients [6.9%] in the placebo group, 2/27 [7.4%] in the 3 mg/kg group [one patient with dementia and one with depression followed by suicide attempt], and 3/27 [11.1%] in the 10 mg/kg cohort [one case of diplopia and unconfirmed aseptic meningitis, one case of diarrhea, and one case of chest pain]). No deaths were reported. The 30 mg/kg cohort was discontinued due to cutaneous hypersensitivity. No improvement was noted in muscle strength, but a trend toward increase in lean body muscle mass using dual-energy x-ray absorptiometry was noted in the 3 mg/kg group (placebo -0.07 ± 0.7, 3 mg/kg 2.4 ± 0.7, p = 0.05). The study was not powered to assess efficacy.e493
Conclusion.
Neutralizing antibody to myostatin (MYO-029) is probably safe and tolerable in patients with BMD and LGMD2A–E and 2I at doses of 1 and 3 mg/kg, although a few serious side effects were noted which require further research. Cutaneous hypersensitivity is noted at 10 and 30 mg/kg doses. There are no data regarding long-term safety. There is probably a trend toward increase in lean body muscle mass, but the study was not powered to assess efficacy (one Class I study).
A 12-month randomized, double-blind, placebo-controlled Class II studye495 evaluated the efficacy of prednisolone 0.35 mg/kg/day for 6 months with crossover to placebo for 6 months in 4 boys with BMD. Isometric muscle strength at 3 months increased by 139% with prednisolone (p < 0.001, corrected for multiple outcomes) but was nonsignificant at 6 months. Three of the 4 patients showed stable or improved muscle strength on prednisolone. Two of the 4 patients improved or stabilized on placebo and 1/4 deteriorated on placebo. The difference was nonsignificant by χ2 test. Although improvements were noted in ankle and wrist dorsiflexors and elbow extensors at 3 months and in knee flexors and neck extensors at 3 and 6 months, these differences were nonsignificant when the authors corrected for multiple outcomes. The study was underpowered to detect a significant improvement or to exclude benefit in other outcomes.e495
Conclusion.
On the basis of one Class II study, prednisolone 0.35 g/kg/day is probably effective to improve isometric muscle strength in patients with BMD after 3 months of treatment.
A Class III studye496 evaluated myoblast transplantation into the tibialis anterior in 3 males with BMD compared with saline injections into the opposite tibialis anterior. Patients were pretreated with cyclosporine A (CyA) for 2 months prior to transplantation and continued it for a year posttransplantation. Force generation in the tibialis anterior was measured bilaterally at baseline, after 2 months of CyA, after myoblast implantation, and after discontinuation of CyA. CyA alone produced a significant bilateral increase in muscle force pretransplantation in one patient, and another patient had significant bilateral increase in tibialis anterior force on CyA posttransplantation, but because the increase was bilateral it was felt to be unlikely to be due to the transplant. None of the biopsies showed dystrophin level changes that could be considered therapeutic.e496
Conclusion.
On the basis of one Class III study, data are inadequate to support or refute the use of myoblast transfer in BMD.
A randomized double-blind Class III studye497 evaluated the effects of self-injected subcutaneous growth hormone (GH) 0.07 mg/kg/week for 3 months in 10 patients with BMD. The cardiomyopathic index (echocardiogram QT:PQ ratio) did not change significantly. The complexity of ventricular premature beats as assessed by the Lown classification system decreased from 4A to 1A in 1/6 patients treated with GH and from 4B to 4A in 1/4 patients treated with placebo. In patients treated with GH, left ventricular mass assessed by echocardiogram increased by 42 g (baseline 150 ± 14, 6 weeks 163 ± 27, 12 weeks 173 ± 27, p <0.05) and reduced nonsignificantly in controls. Relative wall thickness also increased by 12% (baseline 0.23 ± 0.01, 6 and 12 weeks 0.05 ± 0.01, p < 0.05), and end-systolic wall stress dropped by 13% in patients treated with GH (baseline 168 ± 19, 6 weeks 149 ± 14, 12 weeks 146 ± 15, p < 0.05) without change in controls. Plasma levels of brain natriuretic peptide, which were elevated when compared with normal values, decreased by 40% in the active treatment group, whereas no significant changes were detected in the placebo group (GH baseline ± SE 190 ± 60 ng/mL, 12 weeks 114 ± 50, p < 0.05; placebo baseline 205 ± 45, 12 weeks 210 ± 55). Timed functional tests and pulmonary function did not change significantly between groups over 12 weeks. No AEs were noted.e497
Conclusion.
Data are inadequate to support or refute the use of subcutaneous GH injections to improve cardiac and pulmonary function in patients with BMD (one Class III study).
A Class III studye499 evaluated a hand training program consisting of 3 weekly sessions of resistance and stretching exercises (2 self-training, one guided by an occupational therapist) in 12 patients with Welander distal myopathy. There was a one-grade increase in right hand strength on manual muscle testing in 7/12 patients and a 2-grade increase in 1/12. There was also an increase in peak pre–/post–pinch grip as measured by Grippit in 11/12. Range of motion measured with a finger goniometer increased from -470 degrees in the right hand and -790 in the left hand to -260 and -510 degrees, respectively. Self-reported performance of activities of daily living improved in multiple domains. When corrected for multiple outcomes, none of these changes was significant. Mean pre–/post–pinch grip scores, grip strength measured with Grippit, and life satisfaction score changes were also nonsignificant.e499
Conclusion.
On the basis of one Class III study, data are insufficient to support or refute the benefit of a hand exercise program in Welander distal myopathy.
Another Class III studye246 evaluated the effect of endurance training in 9 ambulatory patients with LGMD2I and 9 healthy, sedentary, age-matched controls. The home training program consisted of 30-minute stationary bicycle ergometer exercise sessions at a heart rate corresponding to 65% of maximal oxygen uptake (VO2 max) for 12 weeks. The number of sessions increased progressively for the first 4 weeks to 5 times a week in the last 8 weeks. In the patients, VO2 max and maximal workload (Wmax) increased by 21% and 27%, respectively, at 12 weeks (p < 0.0005). These parameters also increased in the control group, but there was no difference in the absolute increase between groups. Plasma lactate and heart rate did not change before and after training. Self-reported improvement was noted in physical endurance (8/9 patients), leg muscle strength (7/9), and walking distance (6/9). Capillary density increased on the 5 tested muscle biopsies (mean ± SE before: 209 ± 19 mm2, after: 255 ± 30 mm2, p = 0.05). CK levels and mean muscle type II fiber area showed a trend toward increase in 5/9 patients with LGMD2I tested at 12 weeks (CK levels before: 661 ± 154, after: 1,068 ± 298, p = 0.08; type II fiber area before: 7,604 ± 735 mm2, after: 8,982 ± 1,204 mm2, p = 0.09). There were no AEs.e246
Conclusion.
Data are insufficient to support or refute the benefit of endurance training for 12 weeks to improve VO2 max, Wmax, and patient-reported outcomes of leg strength, physical endurance, and walking distance in patients with LGMD2I (one Class III study).
A Class III study by the same authors evaluated endurance training in 11 men with BMD and 7 healthy sedentary men.e344 The exercise protocol was similar to that of the previous study.e246 Six patients continued the protocol 3 times weekly for 12 months. At 12 weeks VO2 max and Wmax improved by 47% and 80%, respectively (p < 0.005), which was 3–4 times higher than the changes in controls. Plasma lactate levels and heart rate were not significantly different. A significant increase in muscle strength of the hip abductors and foot dorsiflexors and plantarflexors was also noted; this was maintained at 12 months (increase in strength [Newtons, mean ± SE]: hip abductors 22 ± 7%, foot dorsiflexors 13 ± 6%, foot plantarflexors 20 ± 4%, p <0.05). At 12 months there was a 40% increase in quadriceps strength as well (hip abduction 13 ± 7%, hip flexion 14 ± 8%, knee extension 40 ± 15%, foot dorsiflexion 25 ± 5%, foot plantarflexion 21 ± 5%, p < 0.05). In one patient LVEF increased from 35% to 50% at 12 months. LVEF did not change in any of the other patients. A “majority” of patients with BMD reported improvement in physical endurance, leg muscle strength, and walking distance after 12 weeks of training. CK levels, lean body mass, and body fat percentage did not change. No changes were noted in muscle fiber diameter or capillary density. In the 6 patients who continued the endurance program for 12 months, the improvement in VO2 max, Wmax, and muscle strength was sustained but did not improve further. In 1/3 patients tested, cardiac EF increased from 35% to 50% at 12 months.e344
Conclusion.
Data are insufficient to determine the effect of 12 weeks of endurance training for improving VO2 max, Wmax, muscle strength, and patient-reported outcomes of physical endurance, leg muscle strength, and walking distance in patients with BMD (one Class III study).
Two Class III studiese462,e465 evaluated the effects of exercise on hIBM3 secondary to a defect in the MYH2 gene. In the first study,e462 8 patients participated in an 8-week home exercise program for 30 minutes/day, 5 days a week on a stationary bicycle. No improvements were seen in any of the outcomes after correction for multiple outcomes. The same authors studied 6 patients from the same families after a similar exercise protocol.e465 Maximal workload increased in all patients (statistical data not provided), and expression of MYH IIx and increase in MYH types I and IIa were noted on postexercise biopsies of the vastus lateralis compared to pre-exercise biopsies (p < 0.05). There was no change in muscle strength of the knee extensors and flexors.
Conclusion.
Data are inadequate to assess the effect of endurance training on maximum workload, muscle strength, or change in the expression of myosin isoforms on muscle biopsy in hIBM3 (2 conflicting Class III studies).
RECOMMENDATIONS FOR THE DIAGNOSIS, EVALUATION, AND MANAGEMENT OF LIMB-GIRDLE AND DISTAL MUSCULAR DYSTROPHIES
The recommendations below encompass 3 major areas: diagnosis, evaluation, and management of muscular dystrophies, including limb-girdle, humeroperoneal, and distal muscular dystrophies. Each recommendation is preceded by clinical context that outlines the evidence, general principles of care, and evidence from related disorders that drive the recommendations.
Key:
EVID: Statements supported directly by the systematically reviewed evidence.
PRIN: An accepted axiom or principle.
RELA: Statements supported by strong evidence not included in the systematic review.
INFER: An inference from one or more of the other statements.
Note: Given the relative paucity of literature directly relevant to LGMDs for some of the clinical questions, some of the recommendations below are based in part on evidence from other neuromuscular disorders, primarily amyotrophic lateral sclerosis (ALS).
Overall management
Clinical context.
Our systematic review has highlighted the medical complexity of caring for patients with muscular dystrophy (EVID). Such patients may develop cardiac, pulmonary, nutritional, and musculoskeletal complications that require the assistance of cardiologists, pulmonologists, orthopedists, physiatrists, physical therapists, occupational therapists, nutritionists, orthotists, and speech pathologists, in addition to neurologists (INFER). In addition, myopathies with a limb-girdle, humeroperoneal, or distal pattern of weakness may be challenging to diagnose (INFER). A specific diagnosis provides patients with “closure,” assists genetic counseling, and directs monitoring for complications and optimal management (PRIN).
Recommendation.
A00. Clinicians should refer patients with suspected muscular dystrophy to neuromuscular centers to optimize the diagnostic evaluation and subsequent management (Level B).
Diagnosis of muscular dystrophies (see also table e-2 and figures 1–5, e-1, and e-2) Clinical context.
Our evidence-based review found that muscular dystrophies have some characteristic features, including a predilection for certain ethnicities, type of inheritance (autosomal dominant, recessive, or X-linked), patterns of weakness (limb-girdle, humeroperoneal, or distal), hypertrophy or atrophy of specific muscle groups, and ancillary characteristics such as scapular winging, level of serum CK, particular EMG abnormalities (e.g., myotonic discharges), cardiac and respiratory involvement, and characteristic features on muscle biopsy (e.g., rimmed vacuoles, myofibrillar myopathy, reducing bodies). Very few features were pathognomonic of a specific disorder; for example, a history of PDB, frontotemporal dementia, or motor neuron disease is pathognomonic of hIBMPFD due to mutations in the gene for VCP. Similarly, the likelihood of genetic diagnosis for the dystroglycanopathies (LGMD2K, LGMD2M, LGMD2N, LGMD2O, and LGMD2P) increases in the presence of abnormalities on brain MRI and abnormal α-dystroglycan immunostaining on muscle biopsy. In most of the other muscular dystrophies reviewed, a constellation of features narrows the differential diagnosis to a few disorders (EVID). The predisposition of certain ethnicities to specific disorders was noted (EVID). However, many disorders are described in several ethnic groups, making it difficult to use ethnicity alone to arrive at a diagnosis. Therefore, in conjunction with specific clinical patterns, certain ethnicities may help to narrow the differential diagnosis and direct confirmatory testing in a proportion of patients (INFER). The limb-girdle and distal muscular dystrophies are presumed to have genetic origins because no plausible environmental cause has been identified. Sporadic cases may be due to autosomal dominant, recessive, or X-linked inheritance (PRIN).
The accurate diagnosis of the muscular dystrophies is important for patients, their families, and for efficient and cost-effective use of medical resources (INFER). Knowing the specific type of muscular dystrophy assists in defining the long-term prognosis, since some dystrophies are more rapidly progressive, involve the cardiorespiratory systems more frequently, or are associated with other disorders (EVID). The identification of these dystrophies through genetic testing will not only inform long-term prognosis but will also assist in directing care more efficiently (e.g., more frequent cardiorespiratory monitoring and prophylactic treatments such as pacer/defibrillator placement for those disorders known to be associated with cardiac involvement) (INFER). Precise identification of the disorder also eliminates the need for repeated testing for an acquired, treatable disorder such as an inflammatory myopathy, because some dystrophies can have inflammation on muscle biopsy, making diagnosis difficult on the basis of routine biopsy findings alone (INFER). In addition, the temptation to try immunosuppressive agents repeatedly, looking for a therapeutic response, is not unusual when there is no diagnosis and the patient is worsening (INFER). This exposes patients to potentially serious side effects of immunosuppressive medications (PRIN). Patients on immunosuppressants need to be monitored at regular intervals, adding logistical difficulties to a population that may have significantly impaired mobility (INFER). Health care costs are also increased by repeated investigations, immunosuppressive treatments, and laboratory monitoring (PRIN). Although establishing a genetic diagnosis is expensive on the front end, the costs of continued investigation for other causes and the risks and expenses associated with empiric trials of immunosuppressants make a strong case for establishing a genetic diagnosis, which often provides patients a sense of “closure” (INFER). Establishing a genetic diagnosis is crucial for genetic counseling of families to inform decision making about having children and for screening of offspring based on the genetics of the disorder (PRIN). Treatment of cardiomyopathy, arrhythmias, and ventilatory failure prolongs life and improves quality of life in patients with other neuromuscular diseases (RELA).e43,e501-e505 The recommendations below also discuss the overall differential diagnosis of LGMD syndromes, including Pompe disease (acid αglucosidase deficiency) and the collagen VI disorders, Ullrich and Bethlem myopathy, which may mimic LGMD or EDMD, respectively. These disorders were not formally reviewed in the evidence since they are not included under LGMD, but they should be considered in the differential diagnosis of LGMD and EDMD.
Recommendations.
A0. For patients with suspected muscular dystrophy, clinicians should use a clinical approach to diagnosis based on the clinical phenotype, including the pattern of muscle involvement, inheritance pattern, age at onset, and associated manifestations (e.g., early contractures, cardiac or respiratory involvement) (Level B).
Limb-girdle pattern of weakness (see figures 3-5).
A1. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal dominant inheritance, cardiomyopathy, respiratory involvement, EMG with myotonic or “pseudomyotonic” discharges (the latter characterized by runs of decrescendo positive sharp wave discharges without the typical waxing and waning of amplitudes and frequencies seen in myotonic discharges), ankle dorsiflexor weakness (foot drop), and muscle biopsy (if performed) showing features of myofibrillar myopathy, clinicians should perform genetic testing for mutations in the genes for desmin (LGMD1E), myotilin (LGMD1A), DNAJB6 (LGMD1D), ZASP, filamin C, αB-crystallin, and titin (Level B).
A2. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal dominant inheritance, rippling muscles, and percussion-induced rapid contractions, clinicians should perform genetic testing for mutations in the caveolin-3 gene (LGMD1C) (Level B).
A3. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal dominant inheritance, early humeroperoneal weakness, contractures (neck, elbows, knee, ankle), and cardiomyopathy, clinicians should perform genetic testing for mutations in the lamin A/C gene (LGMD1B or AD-EDMD) (Level B).
A4. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal dominant inheritance, distal weakness, myotonic discharges on EMG, past or family history of Paget disease, frontotemporal dementia, or motor neuron disease, clinicians should perform genetic testing for mutations in VCP (hIBMPFD) (Level B).
A5. In patients with limb-girdle weakness and suspected muscular dystrophy who either do not have clinical features to suggest a specific form of dystrophy or in whom initial genetic testing is not informative, clinicians should perform muscle biopsy in order to delineate characteristic features that direct further genetic testing (such as immunohistochemistry/immunoblotting for various sarcolemmal proteins, calpain-3, or features of myofibrillar myopathy; see figures 3-5) or to exclude an alternative diagnosis (e.g., a metabolic myopathy, mitochondrial myopathy, congenital myopathy, or inflammatory myopathy) (Level B).
A6. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal recessive inheritance, scapular winging but no calf hypertrophy, and normal cardiorespiratory function, clinicians should perform initial genetic testing for mutations in calpain-3 (LGMD2A). Patients of English, French, Spanish, Italian, Portuguese, or Brazilian descent may have a higher pretest probability of this disorder (Level B).
A7. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal recessive inheritance and calf atrophy and weakness (i.e., inability to stand on toes), clinicians should perform genetic testing for mutations in anoctamin-5 (LGMD2L) or dysferlin (LGMD2B). If the onset of symptoms is in the teens or early 20s or the patient is from Asia, clinicians should assess for dysferlin mutations first and, if negative, test for anoctamin-5 mutations. If the onset of symptoms is in the 30s or later or the patient is of English or northern European ancestry, clinicians should assess for anoctamin-5 mutations first and, if negative, test for dysferlin mutations (Level B).
A8. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal recessive inheritance and muscle biopsy immunohistochemistry showing reduction in α-, β-, γ-, or δ-sarcoglycans, clinicians should perform genetic testing for mutations in the sarcoglycan genes (LGMD2C–2F) (Level B).
A9. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal recessive inheritance who are of Hutterite descent, clinicians should perform genetic testing for mutations in TRIM32 (LGMD2H) (Level B).
A10. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal recessive inheritance, scapular winging, calf hypertrophy, and early cardiorespiratory involvement, clinicians should perform initial genetic testing for mutations in FKRP (LGMD2I). Patients of northern European descent may have a higher pretest probability of this disorder (Level B).
A11. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal recessive inheritance and mental retardation, clinicians should screen for mutations in genes that cause primary or secondary deficiency of α-dystroglycan (LGMD2K, LGMD2M, LGMD2N, LGMD2O, and LGMD2P) (Level B).
A12. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal recessive inheritance and epidermolysis bullosa or pyloric atresia, clinicians should perform genetic testing for mutations in plectin (Level B).
A13. In male patients with limb-girdle weakness and suspected muscular dystrophy with probable X-linked inheritance, clinicians should perform genetic testing for mutations in the dystrophin gene (Duchenne or Becker muscular dystrophy) (Level B).
A14. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal recessive inheritance and no other specific clinical features or in whom muscle biopsy does not inform genetic testing, clinicians should perform dried blood spot test for α-glucosidase (acid maltase) deficiency or Pompe disease (Level B).
A15. In female patients with limb-girdle weakness and suspected muscular dystrophy with probable X-linked inheritance, clinicians should perform genetic testing for dystrophin mutations or perform a muscle biopsy and immunostain for dystrophin to assess for a mosaic pattern of staining. If abnormal immunostaining is present, clinicians should confirm the diagnosis of manifesting carrier of dystrophinopathy with genetic testing for mutations in the dystrophin gene (Level B).
Humeroperoneal weakness (figure e-1).
B1. In patients with humeroperoneal weakness and suspected muscular dystrophy with probable autosomal dominant inheritance, early cardiac involvement, and no joint laxity, clinicians should perform genetic testing for mutations in the lamin A/C gene (AD-EDMD, LGMD1B). If the inheritance pattern is probably X-linked, clinicians should perform genetic testing for mutations in the emerin gene (XR-EDMD) (Level B).
B2. In patients with humeroperoneal weakness and suspected muscular dystrophy with early cardiac involvement and no joint laxity who do not possess mutations in the lamin A/C or emerin gene, clinicians should perform muscle biopsy to delineate characteristic abnormalities that direct further genetic testing (see figure e-1 for muscle biopsy features that direct genetic testing) (Level B).
B3. In patients with humeroperoneal weakness and suspected muscular dystrophy with probable autosomal dominant inheritance, joint laxity, protuberant calcaneus, and no cardiac involvement, clinicians should perform genetic testing for mutations in the collagen VI gene (Bethlem myopathy). If the inheritance pattern is probably autosomal recessive with congenital onset, clinicians should perform genetic testing for mutations in the collagen VI gene (Ullrich myopathy) (Level B).
Distal muscular dystrophy (figure e-2).
C1. In patients with late adult onset of index finger and wrist extensor weakness followed by atrophy and weakness of hand muscles and muscle biopsy showing rimmed vacuoles, clinicians should make a diagnosis of Welander distal myopathy. Patients of Swedish or Finnish descent may have a higher pretest probability of this disorder. Clinicians should confirm the diagnosis with genetic testing for Welander myopathy when testing becomes commercially available (Level B).
C2. In patients with suspected distal muscular dystrophy and probable autosomal recessive inheritance with early onset of calf weakness, clinicians should perform genetic testing for mutations in the anoctamin-5 and dysferlin genes. If the patient is of northern European descent, clinicians should perform initial genetic testing for mutations in the anoctamin-5 gene (LGMD2L) and, if negative, perform genetic testing for mutations in the dysferlin gene (LGMD2B). If the patient is from eastern Asia (Japan, China, Korea), clinicians should perform initial genetic testing for mutations in the dysferlin gene (LGMD2B, Miyoshi myopathy) and, if negative, perform genetic testing for mutations in the anoctamin-5 gene (LGMD2L) (Level B).
C3. In patients with suspected distal muscular dystrophy and probable autosomal recessive inheritance with early onset (<30 years of age) of progressive foot drop who are of Japanese or Middle Eastern Jewish descent, clinicians should perform initial genetic testing for GNE mutations (AR-hIBM) (Level B).
C4. In patients with suspected distal muscular dystrophy without the clinical features in C2 or C3 above, clinicians should perform a muscle biopsy to direct further genetic testing (see figure e-2 for biopsy and clinical features that direct genetic testing) (Level B).
Other diagnostic considerations.
D1. In patients with muscular dystrophy who have proximal as well as distal weakness, clinicians should use specific clinical features (e.g., rippling muscles, cardiomyopathy, atrophy of specific muscle groups, irritability on EMG) and biopsy features (MFM, reduction of emerin immunostaining, presence of rimmed vacuoles) to guide genetic testing, which may include mutations in the genes causing the various forms of MFM (see section on MFM), LGMD2B (dysferlin), LGMD2L (anoctamin-5), LGMD2J (titin), LGMD1C (caveolin-3), and EDMD (emerin and lamin A/C) (Level B).
D2. In patients with suspected muscular dystrophy in whom initial genetic testing, muscle biopsy, and dried blood spot test for Pompe disease do not provide a diagnosis, clinicians may obtain genetic consultation or perform parallel sequencing of targeted exomes, whole-exome sequencing, whole-genome screening, or next-generation sequencing to identify the genetic abnormality (Level C).
Evaluation and medical management of muscular dystrophies
In this section we address monitoring and medical management of complications.
Cardiac involvement.
Clinical context.
Our systematic review reveals that many, though not all, muscular dystrophy subtypes have associated cardiac involvement (EVID). Muscular dystrophy patients with cardiac involvement often do not have symptoms such as chest pain, pedal edema, or palpitations that precede cardiac morbidity or sudden cardiac death. Serious cardiac manifestations in patients with muscular dystrophy are often identified only with cardiology testing (PRIN). The detection and appropriate management of cardiac dysfunction are important to reduce morbidity and mortality (PRIN). Patients with muscular dystrophy often have improved quality of life following appropriate pharmacologic treatment, device placement, or surgical intervention for their cardiac involvement (RELA).e506
Recommendations.
E1. Clinicians should refer newly diagnosed patients with
- LGMD1A, LGMD1B, LGMD1D, LGMD1E, LGMD2C–K, LGMD2M–P, BMD, EDMD, and MFM
- muscular dystrophy without a specific genetic diagnosis for cardiology evaluation, including ECG and structural evaluation (echocardiography or cardiac MRI), even if they are asymptomatic from a cardiac standpoint, to guide appropriate management (Level B).
E1a. If ECG or structural cardiac evaluation (e.g., echocardiography) is abnormal, or if the patient has episodes of syncope, near-syncope, or palpitations, clinicians should order rhythm evaluation (e.g., Holter monitor or event monitor) to guide appropriate management (Level B).
E2. Clinicians should refer muscular dystrophy patients with palpitations or who are found to have symptomatic or asymptomatic tachycardia or arrhythmias for cardiology evaluation (Level B).
E3. Clinicians should refer muscular dystrophy patients with signs or symptoms of cardiac failure for cardiology evaluation (e.g., medical management, left ventricular assist device placement, or cardiac transplantation, as deemed necessary by the cardiologist) to prevent cardiac death (Level B).
Clinical context.
Our systematic review found that muscular dystrophy patients with certain genetic subtypes (LGMD2A, LGMD2B, and LGMD2L) are at very low risk of concomitant cardiac involvement during the course of their disease (EVID). Asymptomatic patients with these muscular dystrophy subtypes would not benefit from cardiac testing. They would only be exposed to the added risk and costs associated with this testing. The quality of life in asymptomatic muscular dystrophy patients with genetic subtypes at very low risk of concomitant cardiac involvement is not improved by cardiology evaluation and testing (INFER).
Recommendation.
E4. It is not obligatory for clinicians to refer patients with LGMD2A, LGMD2B, and LGMD2L for cardiac evaluation unless they develop overt cardiac signs or symptoms (Level B).
Clinical context.
Our systematic review has demonstrated an important risk of symptomatic involvement of both skeletal muscle and cardiac muscle in female carriers of dystrophinopathy and emerinopathy (EVID). About 15% of carriers of dystrophinopathy have cardiac involvement before 15 years of age. This increases to about 45% in patients above 15 years of age. Similarly, about 18% of female carriers of emerinopathy over the age of 60 years have typical ECG abnormalities (EVID). Carriers of these disorders may not have obvious symptoms of skeletal muscle or cardiac involvement (PRIN). The detection and appropriate management of skeletal muscle weakness and cardiac dysfunction is important in order to reduce morbidity and mortality (PRIN). Patients with muscle weakness and cardiac involvement from other disorders often have improved quality of life following appropriate management and treatment of cardiac dysfunction (RELA).e507-e509
Recommendation.
E5. Clinicians should encourage female carriers of dystrophinopathy and emerinopathy to seek evaluation by a neuromuscular specialist and a cardiologist to assess for skeletal muscle and cardiac muscle involvement and to proactively treat cardiac involvement (Level B).
Dysphagia and nutrition.
Clinical context.
Patients with muscular dystrophy may have difficulty receiving adequate oral intake due to dysphagia and/or inability to feed themselves due to excessive arm weakness (EVID). Maintaining adequate nutrition and body weight is important for optimizing strength, function, and quality of life (PRIN). When oral intake is inadequate, other means of maintaining intake, such as gastrostomy or jejunostomy feeding tubes, may be needed to maintain optimal nutrition (PRIN). There is evidence from related conditions (ALS) that maintenance of nutrition and body weight prolongs survival (RELA).e501
Recommendation.
F1. Clinicians should refer muscular dystrophy patients with dysphagia, frequent aspiration, or weight loss for swallowing evaluation and/or gastroenterology evaluation to assess and manage swallowing function and aspiration risk, to teach patients techniques for safe and effective swallowing (e.g., “chin tuck” maneuver, altered food consistencies, etc.), and to consider placement of a gastrostomy/jejunostomy tube for nutritional support (Level B).
Pulmonary complications.
Clinical context.
Our systematic review demonstrates that some forms of muscular dystrophy are associated with oropharyngeal or ventilator muscle weakness and that patients with these forms are at high risk for developing respiratory failure during the course of their disease. Patients with LGMD2B and LGMD2L rarely, if ever, have symptomatic respiratory involvement from their disease (EVID). Patients with respiratory failure from neuromuscular-related weakness often do not have symptoms, such as dyspnea, that precede the onset of respiratory failure. Impending respiratory failure in these patients is often identified only with pulmonary function tests (PRIN). Respiratory failure constitutes a major source of morbidity, interfering with daytime cognitive function and negatively affecting quality of life (PRIN). In addition, ventilatory and oropharyngeal weakness can threaten survival through the risk of upper airway obstruction and/or bellows failure (RELA).e501 Patients with respiratory failure secondary to muscle weakness often have improved quality of life with noninvasive pulmonary ventilation (RELA).e501
Recommendations.
G1. Clinicians should order pulmonary function testing (spirometry and maximal inspiratory/expiratory force in the upright and, if normal, supine positions) or refer for pulmonary evaluation (to identify and treat respiratory insufficiency) in muscular dystrophy patients at the time of diagnosis, or if they develop pulmonary symptoms later in their course (Level B).
G1a. In patients with a known high risk of respiratory failure (e.g., those with LGMD2I or MFM), clinicians should obtain periodic pulmonary function testing (spirometry and maximal inspiratory/expiratory force in the upright position and, if normal, in the supine position) or evaluation by a pulmonologist to identify and treat respiratory insufficiency (Level B).
G2. It is not obligatory for clinicians to refer patients with LGMD2B and LGMD2L for pulmonary evaluation or pulmonary function testing unless they are symptomatic (Level C).
G3. Clinicians should refer muscular dystrophy patients with excessive daytime somnolence, nonrestorative sleep (e.g., frequent nocturnal arousals, morning headaches, excessive daytime fatigue), or respiratory insufficiency based on pulmonary function tests for pulmonary or sleep medicine consultation for consideration of noninvasive ventilation to improve quality of life (Level B).
Cognitive dysfunction and learning disabilities.
Clinical context.
Although cognitive dysfunction, reduced IQ, and learning disabilities are not major factors in most patients with limb-girdle muscular dystrophy, they are noted in a few disorders, such as BMD and those disorders that cause a primary or secondary defect in α-dystroglycan (EVID). Identification and management of these disorders is important to delineate special needs and to provide the resources necessary for these patients to live as normal a life as possible (PRIN).
Recommendation.
H1. In muscular dystrophy patients with symptoms suggestive of cognitive dysfunction or learning disabilities, clinicians may order neuropsychological testing, MRI of the brain, and/or developmental pediatrics consultation to assess for and optimally manage CNS involvement (Level C).
Spinal deformities.
Clinical context.
Our systematic review has revealed the risk of evolving musculoskeletal spine deformities, such as scoliosis, kyphosis, or rigid spine syndrome, in various muscular dystrophies (EVID). These musculoskeletal deformities can result in discomfort and functional impairment, interfering with gait, activities of daily living, and pulmonary function (PRIN). The proper management of musculoskeletal spine deformities is important in order to reduce discomfort, preserve mobility or ability to sit in a wheelchair, and reduce pulmonary complications (RELA).e506
Recommendations.
I1. Clinicians should monitor patients with muscular dystrophy for the development of spinal deformities to prevent resultant complications and preserve function (Level B).
I2. Clinicians should refer muscular dystrophy patients with musculoskeletal spine deformities to an orthopedic spine surgeon for monitoring and surgical intervention if it is deemed necessary in order to maintain normal posture, assist mobility, maintain cardiopulmonary function, and optimize quality of life (Level B).
Osteoporosis.
Clinical context.
Our systematic review did not provide evidence regarding monitoring for osteoporosis with bone density testing (EVID). However, sedentary lifestyle is one risk factor for osteoporosis (PRIN). Therefore, patients with limb-girdle muscular dystrophy causing limited mobility may be prone to osteoporosis (INFER). They are also prone to falls and therefore may be at a high risk for injuries, including fractures (PRIN). The injuries may in turn further limit mobility (PRIN).
Recommendation.
J1. Clinicians may choose to evaluate patients with restricted mobility due to muscular dystrophy with bone density studies for osteoporosis in order to institute timely management and minimize fractures (Level C).
Infection prophylaxis.
Clinical context.
Our systematic review did not provide evidence regarding immunization with pneumococcal vaccination or annual influenza vaccination (EVID). Given the underlying respiratory muscle weakness or spinal deformities in some subtypes of muscular dystrophy, prevention of respiratory infections is important in order to avoid complications, such as respiratory failure, requiring ventilator support (RELA).e510-e513 The Centers for Disease Control and Prevention (CDC) recommends pneumococcal polysaccharide vaccine (PPSV23) for “all adults aged 65 years and older; adults younger than age 65 years with chronic lung disease (including chronic obstructive pulmonary disease, emphysema, and asthma); chronic cardiovascular diseases; diabetes mellitus; chronic renal failure; nephrotic syndrome; chronic liver disease (including cirrhosis); alcoholism; cochlear implants; cerebrospinal fluid leaks; immunocompromising conditions; and functional or anatomic asplenia;…residents of nursing homes or long-term care facilities; and adults who smoke cigarettes” (PRIN).e514 Patients with limb-girdle muscular dystrophy may be considered as having a chronic illness, may have cardiorespiratory involvement, and may be residents of longterm care facilities (INFER). Influenza vaccine is recommended annually for all persons over 6 months of age (PRIN).
Recommendation.
K1. Clinicians should recommend pneumococcal polysaccharide vaccine (PPSV23) as per the CDC schedulee514 and annual influenza vaccine to patients with muscular dystrophy in order to prevent respiratory complications of pneumococcal pneumonia and influenza (Level B).
Rehabilitative management and treatment of muscular dystrophies
Clinical rehabilitative management.
Clinical context.
Our evidence-based review of the literature on rehabilitative management of muscular dystrophies (LGMD, EDMD, and distal myopathy) consisted primarily of single Class III studies. Thus, the currently available data are not adequate to properly assess the effect of any rehabilitation modality (endurance and strength training, bracing, and assistive devices, including new computer-based technology) (EVID). Large well-designed clinical trials are therefore required to evaluate the role of rehabilitative treatments for these disorders (INFER). However, the principles of the long-term management of patients with these disorders must emphasize maintaining mobility and functional independence for as long as possible, with a focus on maximizing quality of life. The prevention and management of comorbidities, both expected and acquired, is a major part of such management. These comorbidities would include joint contractures, scoliosis, osteoporosis, dysphagia, and restrictive lung disease (expected), as well as obesity, metabolic syndrome, and stress fractures (acquired). Patient-centered, proactive, and collaborative decision making (including all relevant team members) is important, taking into account the patient’s wishes and family and social circumstances. An important aspect of ongoing management includes proactively preparing patients with muscular dystrophy and their families for the long-term consequences of muscular dystrophies and engaging in discussions regarding end-of-life care. This helps patients come to terms with their condition and prepare for the expected complications of their form of muscular dystrophy and avoids the need for hasty decisions made in the throes of a medical crisis (PRIN). There is evidence from studies in other neuromuscular diseases, including ALS, that a multidisciplinary approach is the most effective way to deliver care (RELA).e515 This model is endorsed by the Muscular Dystrophy Association, which also sponsors these types of clinics. This level of care usually occurs at a tertiary or academic-based medical center, with clinics designed specifically to care for patients with muscular dystrophy and other neuromuscular disorders. The primary clinic team members usually include a neurologist and a physiatrist, along with physical and occupational therapists. Neurogeneticists, pulmonologists, cardiologists, gastroenterologists, orthopedic surgeons, and speech-language pathologists are part of the team. Rehabilitation psychologists, social workers, and vocational rehabilitation counselors can also be valuable members of the team. Once the diagnosis is confirmed, the rehabilitation team, under the direction of a physician (preferably a neuromuscular-trained specialist), can manage clinical problems long-term. Ideally, this would include complete functional assessments using reliable, standardized, reproducible measures in order to quantify a patient’s physical and psychosocial performance at any given point in the disease process. This not only helps monitor a patient’s overall level of health but also facilitates evaluation for possible enrollment in a clinical trial. With the loss of mobility will come the need for progressively more assistance with activities of daily living, as well as potential medical complications (PRIN). When writing therapy prescriptions, clinicians should be aware of the current allowances from payers for outpatient physical therapy. Patients with muscular dystrophy should see physical and occupational therapists who are experienced in treating these disorders. These therapists often practice in a tertiary care, medical center–based setting (PRIN).
Recommendations.
L1. Clinicians should refer patients with muscular dystrophy to a clinic that has access to multiple specialties (e.g., physical therapy, occupational therapy, respiratory therapy, speech and swallowing therapy, cardiology, pulmonology, orthopedics, and genetics) designed specifically to care for patients with muscular dystrophy and other neuromuscular disorders in order to provide efficient and effective long-term care (Level B).
L2. Clinicians might discuss opportunities for participation in clinical trials, if available, with muscular dystrophy patients (Level C).
L3. Clinicians should recommend that patients with muscular dystrophy have periodic assessments by a physical and occupational therapist for symptomatic and preventive screening (Level B).
L4. While respecting and protecting patient autonomy, clinicians should proactively anticipate and facilitate patient and family decision making as the disease progresses, including decisions regarding loss of mobility, need for assistance with activities of daily living, medical complications, and end-of-life care (Level B).
L5. For patients with muscular dystrophy, clinicians should prescribe physical and occupational therapy, as well as bracing and assistive devices that are adapted specifically to the patient’s deficiencies and contractures, in order to preserve mobility and function and prevent contractures (Level B).
Strength training and aerobic exercise training.
Clinical context.
As mentioned earlier, our evidence-based review of the literature on rehabilitation management of muscular dystrophies (LGMD, EDMD, and distal myopathy) consisted primarily of single Class III studies. Thus, the currently available data are not adequate to properly assess the effect of any rehabilitation modality (endurance and strength training, bracing, and assistive devices, including new computer-based technology) (EVID). Despite inadequate research in this area, the available evidence suggests that this population would benefit from both strengthening and aerobic fitness training programs. Due to the muscle degeneration in muscular dystrophy, there may be some risk of exercise-induced muscle damage and subsequent overwork weakness following supramaximal, high-intensity exercise. Overwork weakness is defined as a prolonged decrease in absolute muscle strength and endurance following strenuous or excessive exercise. It is often accompanied by extreme delayed onset muscle soreness, peaking 1–5 days after exercise and possibly inducing myoglobinuria. Clinicians need to be prudent in their recommendations, encouraging alternating periods of physical activity and scheduled rest. Clinicians should also be aware that true overwork weakness has not been demonstrated in any trial of exercise done in this population to date. Future investigations should focus on the primary symptom of fatigue and quantify changes in the ability to work and participate in physical activities as outcome measures of an exercise program. All forms of physical exercise should therefore be prescribed cautiously, using a common sense approach (PRIN). There have been several randomized or quasi-randomized controlled trials comparing strength training programs, aerobic exercise programs, or both to non-training controls in patients with a variety of neuromuscular disorders (RELA).e516-e518 On the basis of this literature, both strength training and aerobic exercise programs appear to be safe, without any notable deleterious effects. However, limitations in the design of these trials prevent any conclusions regarding possible benefit (EVID, RELA).
Recommendations.
M1. Clinicians may advise patients with muscular dystrophy that aerobic exercise combined with a supervised submaximal strength training program is probably safe (Level C).
M2. Clinicians may advise patients with muscular dystrophy that gentle, low-impact aerobic exercise (swimming, stationary bicycling) improves cardiovascular performance, increases muscle efficiency, and lessens fatigue (Level C).
M3. Clinicians may counsel patients with muscular dystrophy to hydrate adequately, not to exercise to exhaustion, and to avoid supramaximal, high-intensity exercise (Level C).
M4. Clinicians should educate patients with muscular dystrophy who are participating in an exercise program about the warning signs of overwork weakness and myoglobinuria, which include feeling weaker rather than stronger within 30 minutes after exercise, excessive muscle soreness 24–48 hours following exercise, severe muscle cramping, heaviness in the extremities, and prolonged shortness of breath (Level B).
Medical treatments.
Clinical context.
Our systematic review of treatments available for LGMD revealed that adeno-associated virus gene transfer increased the expression of the γ-sarcoglycan and α-sarcoglycan genes in the injected muscle for 1 and 6 months, respectivelye494,e498,e500 (EVID). These are small proof-of-concept studies. Despite evidence of increased expression of the target protein at the site of injection, effects on the clinical course of the disorder and the long-term side effects of this treatment are yet to be determined (INFER).
Our systematic review found that neutralizing antibody to myostatin (MYO-029) is probably safe and tolerable in patients with BMD and LGMD2A–E and 2I at doses of 1 and 3 mg/kg, although a few serious side effects were noted which require further research. Cutaneous hypersensitivity is noted at 10 and 30 mg/kg doses. There are no data regarding long-term safety. There is probably a trend toward increase in lean body muscle mass, but the study was not powered to assess efficacye493 (EVID).
Our systematic review found only one study evaluating the effect of myoblast transplantatione496 and one study evaluating the effects of subcutaneous growth hormone injections in BMD,e497 both with inconclusive results.
Recommendations.
N1. Clinicians should not offer patients with LGMD gene therapy outside of a research study designed to determine the efficacy and safety of the treatment (Level R).
N2. Clinicians should not offer patients with LGMD neutralizing antibody to myostatin outside of a research study designed to determine the efficacy and safety of the treatment (Level R).
N3. Clinicians should not offer patients with BMD myoblast transplantation or subcutaneous growth hormone injections outside of a research study designed to determine the efficacy and safety of the treatment (Level R).
RECOMMENDATIONS FOR FUTURE RESEARCH
As the category of LGMDs expands rapidly with advances in molecular diagnostics and new disorders due to specific gene defects are identified, there is need for research in the following areas.
- Larger prospective, long-term, population-based studies are required to establish the prevalence of these rare disorders, identify the ethnic populations among which they are most prevalent, and evaluate their long-term course, including the incidence of cardiorespiratory complications.
- Ongoing studies of genotype/phenotype correlation are needed to help establish phenotypic patterns based on genotype and to describe the various phenotypes that are caused by one genotype.
- The optimal management of cardiorespiratory complications (e.g., frequency and types of screening, effective treatments) should be evaluated.
- Well-designed studies of the effectiveness of exercise programs, physical therapy, and endurance training are needed.
- Studies of other treatments should be conducted, including symptomatic treatments such as the effect of orthotics for contractures (nonsurgical/surgical) on mobility and quality of life, as well as specific disease-modifying treatments such as gene therapy and stem cell therapy.
- Preliminary data suggest that corticosteroids may be effective in α-dystroglycanopathies.
This finding needs to be replicated in larger, controlled studies.
DISCLAIMER
This statement is provided as an educational service of the American Academy of Neurology and American Association of Neuromuscular & Electrodiagnostic Medicine. It is based on an assessment of current scientific and clinical information. It is not intended to include all possible proper methods of care for a particular neurologic problem or all legitimate criteria for choosing to use a specific procedure. Neither is it intended to exclude any reasonable alternative methodologies. The AAN and AANEM recognize that specific patient care decisions are the prerogative of the patient and the physician caring for the patient, based on all of the circumstances involved. The clinical context section is made available in order to place the evidence-based guideline(s) into perspective with current practice habits and challenges. Formal practice recommendations are not intended to replace clinical judgment.
CONFLICT OF INTEREST
The American Academy of Neurology and American Association of Neuromuscular & Electrodiagnostic Medicine are committed to producing independent, critical, and truthful clinical practice guidelines (CPGs). Significant efforts are made to minimize the potential for conflicts of interest to influence the recommendations of this CPG. To the extent possible, the AAN and AANEM keep separate those who have a financial stake in the success or failure of the products appraised in the CPGs and the developers of the guidelines. Conflict of interest forms were obtained from all authors and reviewed by an oversight committee prior to project initiation. AAN and AANEM limit the participation of authors with substantial conflicts of interest. The AAN and AANEM forbid commercial participation in, or funding of, guideline projects. Drafts of the guideline have been reviewed by at least three AAN committees, at least one AANEM committee, a network of neurologists, Neurology peer reviewers, and representatives from related fields. The AAN Guideline Author Conflict of Interest Policy can be viewed at www.aan.com. For complete information on this process, access the 2004 AAN process manual.e7



Abbreviations: AD = autosomal dominant; AR = autosomal recessive; EDMD = Emery-Dreifuss muscular dystrophy; FTD = frontotemporal dementia; hIBM = hereditary inclusion body myopathy; XR = X-linked recessive.
*Described after this systematic review was performed and hence not discussed in this guideline.
Appendix e-1:Mission statement of GDS
The mission of the GDS is to prioritize, develop, and publish evidence-based guidelines related to the diagnosis, treatment, and prognosis of neurological disorders.
The GDS is committed to using the most rigorous methods available within our budget, in collaboration with other available AAN resources, to most efficiently accomplish this mission.
Appendix e-2: 2013–2015 Guideline Development Subcommittee (GDS) members
Cynthia Harden, MD (Chair); Steven R. Messé, MD, FAAN (Vice-Chair); Richard L. Barbano, MD, PhD, FAAN; Jane Chan, MD, FAAN; Diane Donley, MD; Terry Fife, MD, FAAN; Jeffrey Fletcher, MD; Michael Haboubi, MD; John J. Halperin, MD, FAAN; Cheryl Jaigobin, MD; Andres M. Kanner, MD; Jason Lazarou, MD; David Michelson, MD; Pushpa Narayanaswami, MD, MBBS; Maryam Oskoui, MD; Tamara Pringsheim, MD; Alexander Rae-Grant, MD; Kevin Sheth, MD, FAHA; Kelly Sullivan, PhD; Theresa A. Zesiewicz, MD, FAAN; Jonathan P. Hosey, MD, FAAN (Ex-Officio); Stephen Ashwal, MD, FAAN (Ex-Officio); Deborah Hirtz, MD, FAAN (Ex-Officio); Jacqueline French, MD, FAAN (Ex-Officio)
Appendix e-3: AANEM Practice Issues Review Panel (PIRP) members
Yuen T. So, MD, PhD (Co-Chair); Williams S. David, MD, PhD (Co-Chair); Paul E. Barkhaus, MD; Earl J. Craig, MD; Prabhu D. Emmady, MD; Kenneth J. Gaines, MD; James F. Howard, MD; Atul T. Patel, MD; Bharathi Swaminathan, MD; Darrell T. Thomas, MD; Gil I. Wolfe, MD
Appendix e-4: Complete search strategy
Ovid Medline
1950 to December Week 3 2010 # Searches Results Search Type
- muscular dystrophies/ and (lbmd* or (limb adj girdle) or distal or becker or emery or dreifuss or markesberry or griggs or udd or minoka or myoshi or laing or williams or welander).mp. [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 1974 Advanced
- muscular dystrophy, limb girdle/ or ((muscle diseases/ or myopath*.mp.) and distal.mp.) [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 1080 Advanced
- 1 or 2 2829 Advanced
- exp muscular dystrophies/ 18431 Advanced
- laminin*.mp. or lmna protein, human/ or myot protein, human.mp. or calpain.mp. or calpainopath*.mp. or capn3 protein, human/ or dysferlin*.mp. or dysf, protein, human/ [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 24315 Advanced
- exp dystrophin associated proteins/ or sarcoglycan*.mp. or dystroglycans*.mp. or telethonin.mp. or tcap protein, human.mp. [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 1781 Advanced
- (trim32* or fukutin*).mp. or fkrp protein, human/ or titin.mp. or tcap protein, human.mp. or tcap*.mp. or pomt*.mp. or ano5*.mp. or anoctamin.mp. or pomgnt*.mp. or desmin.mp. or alphacrystallin b chain.mp. or ldb3 protein, human.mp. or zasp.mp. [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 8612 Advanced
- (bag3* or filamin or fhli protein, human or emerin or emd protein, human or resprin* or myosin heavy chain or myhct or nvl protein, human or matrin 3 or cavin).mp. [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 7771 Advanced
- 4 and (5 or 6 or 7 or 8) 1638 Advanced
- exp muscular dystrophies/ep, ge, cl, bl, pa 10239 Advanced
- 3 and 10 1903 Advanced
- and (exp molecular diagnostic techniques/ or genetic testing.mp. or exp genetic techniques/) [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 1205 Advanced
- 3 and (exp polymerase chain reaction/ or heterozygote detection/ or exp nucleic amplification techniques/ or exp dna probes/ or ("x" adj linked).mp.) [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 617 Advanced
- 9 or 10 or 12 or 13 10712 Advanced
- 14 not case reports/ 8685 Advanced
- ..l/ 15 hu=y and yr=1987-2010 5546 Advanced
- Muscular Dystrophy, Facioscapulohumeral/ 326 Advanced
- 16 not (17 or fascioscap*.mp.) [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 5364 Advanced
- 3 and 18 1373 Advanced
- 1 or 2 or (exp *muscular dystrophies/ and 14) 10114 Advanced
- ..l/ 20 hu=y and yr=1987-2010 6839 Advanced
- 21 and (creatine kinase/an, bl, du or exp muscles/pa, pp or biops*.mp. or exp imaging, diagnostic/ or physical examination/) [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 2727 Advanced
- [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 690821 Advanced
- exp Cardiovascular Physiological Processes/ 416537 Advanced
- 21 and (23 or 24) 409 Advanced
- muscle weakness or winging*).mp. or ambulation/ or quality of life/ or activities of daily living/ or muscular atrophy/ or exp muscles/pa, pp or exp physical therapy modalities/ or muscle strength.mp. or hand strength/ or hand grip/ or grip strenth.mp. or exercise*.mp. or physical endurance/ [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 502739 Advanced
- 21 and 26 2122 Advanced
- exp Scoliosis/ or exp Braces/ 14674 Advanced
- (absorptiometry, photon or densitometry*).mp. or bone density/ or exp bone resorption/ or exp bone disease, metabolic/ [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 109052 Advanced
- 21 and (28 or 29) 44 Advanced
- exp respiratory insufficiency/ or exp respiratory function tests/ or swallow*.mp. or dysphagia.mp. or exp vocal disorders/ or polysomnography/ or exp respiration, artificial/ or tracheostomy/ or capnography.mp. or hypercapnia.mp. [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 281320 Advanced
- exp Complementary Therapies/ 142979 Advanced
- exp Gene Therapy/ 31909 Advanced
- 21 and (31 or 32 or 33) 467 Advanced
- 22 or 25 or 27 or 30 or 34 3421 Advanced
- limit 35 to (clinical trial, all or clinical trial, phase i or clinical trial, phase ii or clinical trial, phase iii or clinical trial, phase iv or clinical trial or comparative study or controlled clinical trial or meta analysis or multicenter study or practice guideline or randomized controlled trial) 348 Advanced
- 35 and ((cohort* or observation* or population* or retrospective* or prospective*).mp. or review.pt. or (case adj series).mp. or (case adj controlled).mp.) [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 863 Advanced
- 36 or 37 1134 Advanced
- limit 38 to yr="1987 - 2012" 266 Advanced
PubMed
(LGMD* OR “limb girdle muscular dystrophy” OR distal myopathies[mesh] OR “distal muscular dystrophy” OR “Becker muscular dystrophy” OR “Emery Dreifuss muscular dystrophy” OR “inclusion body myositis” OR “inclusion body myopathy” OR muscular dystrophy, limb girdle[mesh] OR muscular diseases[mesh:noexp] OR muscle weakness[mesh] OR muscular dystrophies[mesh:noexp]) AND (genetics[sh] OR sarcotubular OR dnajb6 OR kelch OR myotilin OR “lamin a/c” OR “caveolin-3” OR “calpain-3” OR dysferlin OR sarcoglycan OR telethonin OR TRIM32 OR FKRP OR Titin OR “o-mannosyltransferase-1” OR POMT* OR fukutin OR anoctamin OR ANO5 OR “o-mannose- beta 1,2-N-Acetylglucosaminyl transferase” OR POMTGnT1 OR “O-mannosyltransferase-2” OR POMT OR desmin OR “Alpha b Crystallin” OR ZASP OR “Bag-3” OR filamin* OR FHL1* OR fhl OR lim domain proteins[mesh] OR emerin OR nesprin OR “Markesbery-Griggs” OR Udd OR Nonaka OR Miyoshi OR Laing OR Williams OR Welander OR “myosin heavy chain” OR “MyHC 7” OR VCP OR “Valosin containing protein” OR “Matrin 3” OR GNE OR epimerase OR Cavin OR selenoprotein OR SEPN1 OR VCPDM OR nebulin OR ”rigid spine” OR dystroglycan OR “transmembrane protein 43” OR tmem* OR LUMA) NOT muscular dystrophy, duchenne[majr] Cochrane, Embase, Web of Science
Ovid MEDLINE(R) In-Process & Other Non-Indexed Citations and Ovid MEDLINE(R) 1946 to Present # Searches Results Search Type
- (lgmd* or "limb girdle muscular dystrophy").mp. or exp distal myopathies/ or "distal muscular dystrophy".mp. or "becker muscular dystrophy".mp. or "emery dreifuss muscular dystrophy".mp. or "inclusion body myositis".mp. or "inclusion body myopathy".mp. or exp muscular dystrophy, limb girdle/ or muscule weakness/ or muscular dystrophy/ or muscular dystrophies/ [mp=title, abstract, original title, name of substance word, subject heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] 15307 Advanced
- 1 and ge.fs. 6968 Advanced
- 1 and (sarcotubular or dnajb6 or kelch or myotilin or "lamin a/c" or "caveolin-3" or "calpain3").mp. [mp=title, abstract, original title, name of substance word, subject heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] 498 Advanced
- 1 and (dysferlin or sarcoglycan or telethonin or trim32 or fkrp or titin or "omannosyltransferase-1" or pomt* or fukutin or anoctamin or ano5 or "o-mannose-beta1,2nacetylglucosaminyltransferase" or pomtgnt1).mp. [mp=title, abstract, original title, name of substance word, subject heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] 921 Advanced
- 1 and ((desmin or "alpha b crystallin" or zasp or "bag-3" or filamin* of fhl1* or fhl).mp. or exp lim domain proteins/) [mp=title, abstract, original title, name of substance word, subject heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] 129 Advanced
- 1 and (emerin or nesprin or "markesbery griggs" or udd or nonaka or miyoshi or laing or williams or welander or "myosin heavy chain" or myhc7 or myhc or vcp or "valosin containing protein" or "matrin 3" or gne or epimerase or cavin or selenoprotein* or sepn1 or vcpdm or nebulin or "rigid spine" or dystroglycan or "transmembrane protein 43" or tmem* or luma).mp. [mp=title, abstract, original title, name of substance word, subject heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] 1182 Advanced
- 2 or 3 or 4 or 5 or 6 7393 Advanced
- 7 not *muscular dystrophy, duchenne/ 7167 Advanced
- exp molecular diagnostic techniques/ or "genetic testing".mp. or exp genetic techniques/ or exp polymerase chain reaction/ or heterozygote detection/ or exp nucleic amplications techniques/ or exp dna probes/ or ("x" adj linked).mp. [mp=title, abstract, original title, name of substance word, subject heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] 1418455 Advanced
- 1 and 9 4060 Advanced
- 7 or 10 7702 Advanced
- 11 not *muscular dystrophy, duchenne/ 7466 Advanced
- 12 not (muscular dystrophy, fascioscapulohumeral/ or fascioscap*.mp.) [mp=title, abstract, original title, name of substance word, subject heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] 7461 Advanced
- limit 13 to (english language and humans and yr="1987 - 2012") 4567 Advanced
- 14 and (creatine kinase/an, bl, du or exp muscles/pa, pp or biops*.mp. or exp imaging, diagnostic/ or physical examination/) [mp=title, abstract, original title, name of substance word, subject heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] 1833 Advanced
- exp heart diseases/di, pa, ep, mo, pp, co or exp heart function tests/ or exp electrocardiography/ or exp echocardiography/ or exp assisted circulation/ or pacemaker, artificial/ or cardiac pacing, artificial.mp. or exp heart transplantation/ 750885 Advanced
- exp cardiovascular physiological processes/ 446803 Advanced
- 14 and (16 or 17) 211 Advanced
- (muscle weakness or winging*).mp. or ambulation/ or quality of life/ or activities of daily living/ or muscular atrophy/ or exp muscles/pa, pp or exp physical therapy modalities/ or muscle strength/ or hand strength/ or hand grip/ or "grip strength".mp. or exercise*.mp. or physical endurance/ [mp=title, abstract, original title, name of substance word, subject heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] 572576 Advanced
- 14 and 19 1405 Advanced
- exp scoliosis/ or exp braces/ or bone density/ or exp bone resorption/ or absorptiometry, photon/ or densitomet*.mp. or exp bone diseases, metabolic/ [mp=title, abstract, original title, name of substance word, subject heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] 140311 Advanced
- 14 and 21 40 Advanced
- exp respiratory insufficiency/ or exp respiratory function tests/ or swallow*.mp. or dysphagia.mp. or exp vocal disorders/ or polysomnography/ or exp respiration, artificial/ or tracheostomy/ or capnography.mp. or hypercapn*.mp. or exp deglutition disorders/ [mp=title, abstract, original title, name of substance word, subject heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] 332065 Advanced
- 14 and 23 83 Advanced
- 14 and (exp complementary therapies/ or exp gene therapy/) 185 Advanced
- 15 or 18 or 20 or 22 or 24 or 25 2235 Advanced
Updated Ovid Medline
1950 to December Week 3 2010 # Searches Results Search Type 1 muscular dystrophies/ and (lbmd* or (limb adj girdle) or distal or becker or emery or dreifuss or markesberry or griggs or udd or minoka or myoshi or laing or williams or welander).mp. [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 1974 Advanced 2 muscular dystrophy, limb girdle/ or ((muscle diseases/ or myopath*.mp.) and distal.mp.) [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 1080 Advanced 3 1 or 2 2829 Advanced 4 exp muscular dystrophies/ 18431 Advanced 5 laminin*.mp. or lmna protein, human/ or myot protein, human.mp. or calpain.mp. or calpainopath*.mp. or capn3 protein, human/ or dysferlin*.mp. or dysf, protein, human/ [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 24315 Advanced 6 exp dystrophin associated proteins/ or sarcoglycan*.mp. or dystroglycans*.mp. or telethonin.mp. or tcap protein, human.mp. [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 1781 Advanced 7 (trim32* or fukutin*).mp. or fkrp protein, human/ or titin.mp. or tcap protein, human.mp. or tcap*.mp. or pomt*.mp. or ano5*.mp. or anoctamin.mp. or pomgnt*.mp. or desmin.mp. or alphacrystallin b chain.mp. or ldb3 protein, human.mp. or zasp.mp. [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 8612 Advanced 8 (bag3* or filamin or fhli protein, human or emerin or emd protein, human or resprin* or myosin heavy chain or myhct or nvl protein, human or matrin 3 or cavin).mp. [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 7771 Advanced 9 4 and (5 or 6 or 7 or 8) 1638 Advanced 10 exp muscular dystrophies/ep, ge, cl, bl, pa 10239 Advanced 11 3 and 10 1903 Advanced 12 3 and (exp molecular diagnostic techniques/ or genetic testing.mp. or exp genetic techniques/) [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 1205 Advanced 13 3 and (exp polymerase chain reaction/ or heterozygote detection/ or exp nucleic amplification techniques/ or exp dna probes/ or ("x" adj linked).mp.) [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 617 Advanced 14 9 or 10 or 12 or 13 10712 Advanced 15 14 not case reports/ 8685 Advanced 16 ..l/ 15 hu=y and yr=1987-2010 5546 Advanced 17 Muscular Dystrophy, Facioscapulohumeral/ 326 Advanced 18 16 not (17 or fascioscap*.mp.) [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 5364 Advanced 19 3 and 18 1373 Advanced 20 1 or 2 or (exp *muscular dystrophies/ and 14) 10114 Advanced 21 ..l/ 20 hu=y and yr=1987-2010 6839 Advanced 22 21 and (creatine kinase/an, bl, du or exp muscles/pa, pp or biops*.mp. or exp imaging, diagnostic/ or physical examination/) [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 2727 Advanced 23 exp heart diseases/di, pa, ep, mo, pp, co or exp heart function tests/ or exp electrocardiography/ or exp echocardiography/ or exp assisted circulation/ or pacemaker, artificial/ or cardiac pacing, artificial.mp. or exp heart transplantation/ [mp=title, original title, abstract, name of substance
word, subject heading word, unique identifier] 690821 Advanced 24 exp Cardiovascular Physiological Processes/ 416537 Advanced 25 21 and (23 or 24) 409 Advanced 26 (muscle weakness or winging*).mp. or ambulation/ or quality of life/ or activities of daily living/ or muscular atrophy/ or exp muscles/pa, pp or exp physical therapy modalities/ or muscle strength.mp. or hand strength/ or hand grip/ or grip strenth.mp. or exercise*.mp. or physical endurance/ [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 502739 Advanced 27 21 and 26 2122 Advanced 28 exp Scoliosis/ or exp Braces/ 14674 Advanced 29
(absorptiometry, photon or densitometry*).mp. or bone density/ or exp bone resorption/ or exp bone disease, metabolic/ [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 109052 Advanced 30 and (28 or 29) 44 Advanced 31 exp respiratory insufficiency/ or exp respiratory function tests/ or swallow*.mp. or dysphagia.mp. or exp vocal disorders/ or polysomnography/ or exp respiration, artificial/ or tracheostomy/ or capnography.mp. or hypercapnia.mp. [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 281320 Advanced 32 exp Complementary Therapies/ 142979 Advanced 33 exp Gene Therapy/ 31909 Advanced 34 21 and (31 or 32 or 33) 467 Advanced 35 or 25 or 27 or 30 or 34 3421 Advanced 36 limit 35 to (clinical trial, all or clinical trial, phase i or clinical trial, phase ii or clinical trial, phase iii or clinical trial, phase iv or clinical trial or comparative study or controlled clinical trial or meta analysis or multicenter study or practice guideline or randomized controlled trial) 348 Advanced 37 and ((cohort* or observation* or population* or retrospective* or prospective*).mp. or review.pt. or (case adj series).mp. or (case adj controlled).mp.) [mp=title, original title, abstract, name of substance word, subject heading word, unique identifier] 863 Advanced 38 or 37 1134 Advanced 39 limit 38 to yr="1987 - 1995" 266 Advanced
Appendix e-5: Classification of Evidence Schemes Screening
Class I: A statistical, population-based sample of patients studied at a uniform point in time (usually early) during the course of the condition. All patients undergo the intervention of interest. The outcome, if not objective, is determined in an evaluation that is masked to the patients’ clinical presentations.
Class II: A statistical, non-referral-clinic-based sample of patients studied at a uniform point in time (usually early) during the course of the condition. Most patients undergo the intervention of interest. The outcome, if not objective, is determined in an evaluation that is masked to the patients’ clinical presentations.
Class III: A sample of patients studied during the course of the condition. Some patients undergo the intervention of interest. The outcome, if not objective, is determined in an evaluation by someone other than the treating physician.
Class IV: Studies not meeting Class I, II or III criteria including consensus, expert opinion or a case report
Therapy
Class I: Prospective, randomized, controlled clinical trial with masked outcome assessment, in a representative population. The following are required:
- Concealed allocation
- primary outcome(s) clearly defined
- exclusion/inclusion criteria clearly defined
- adequate accounting for drop-outs and cross-overs with numbers sufficiently low to have minimal potential for bias
- relevant baseline characteristics are presented and substantially equivalent among treatment groups or there is appropriate statistical adjustment for differences.
Class II: Prospective matched group cohort study in a representative population with masked outcome assessment that meets a-e above OR a RCT in a representative population that lacks one criteria a-d.
Class III: All other controlled trials (including well-defined natural history controls or patients serving as own controls) in a representative population, where outcome is independently assessed, or independently derived by objective outcome measurement.
Class IV: Evidence from uncontrolled studies, case series, case reports, or expert opinion.
Appendix e-6: Clinical Contextual Profiles Determining Levels of Obligation for Recommendations
Diagnosis of Muscular Dystrophies
A00. Clinicians should refer patients with suspected muscular dystrophy to neuromuscular centers to optimize the diagnostic evaluation and subsequent management (Level B).
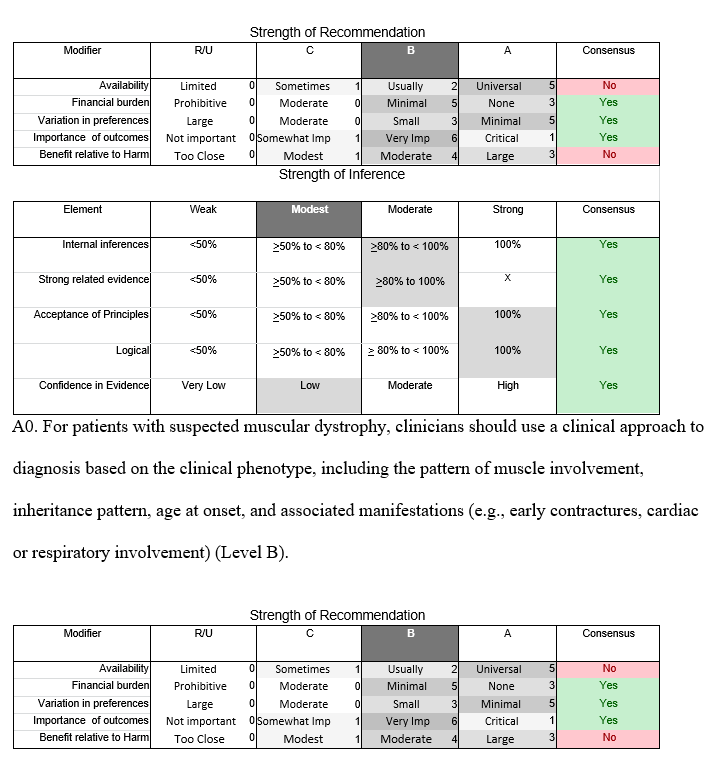

Limb-girdle pattern of weakness (figures 3 to 5).
A1. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal dominant inheritance, cardiomyopathy, respiratory involvement, EMG with myotonic or “pseudomyotonic” discharges (the latter characterized by runs of decrescendo positive sharp wave discharges without the typical waxing and waning in amplitude and frequency seen in myotonic discharges), ankle dorsiflexor weakness (foot drop), and muscle biopsy (if performed) showing features of myofibrillar myopathy, clinicians should perform genetic testing for mutations in the genes for desmin (LGMD1E), myotilin (LGMD1A), DNAJB6 (LGMD1D), ZASP, filamin C, αB-crystallin, and titin (Level B).
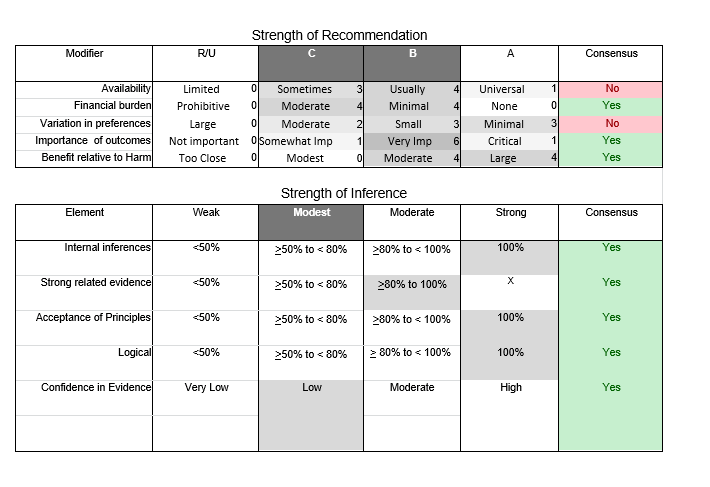
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level B using informal consensus.
A2. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal dominant inheritance, rippling muscles, and percussion-induced rapid contractions, clinicians should perform genetic testing for mutations in the caveolin-3 gene (LGMD1C) (Level B).
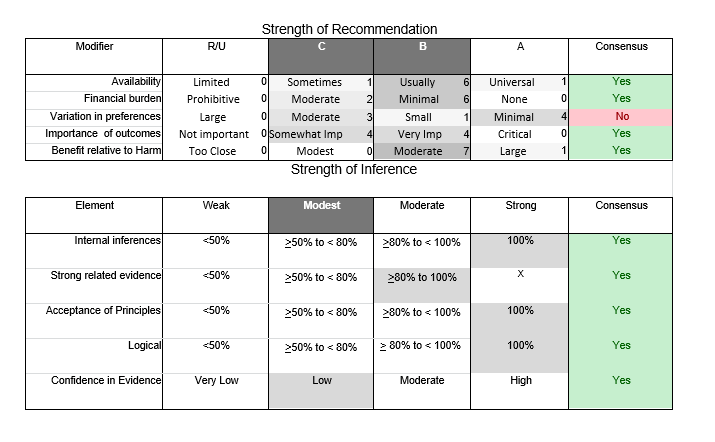
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level B using informal consensus.
A3. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal dominant inheritance, early humeroperoneal weakness, contractures (neck, elbows, knee, ankle), and cardiomyopathy, clinicians should perform genetic testing for mutations in the lamin A/C gene (LGMD1B or AD-EDMD) (Level B).
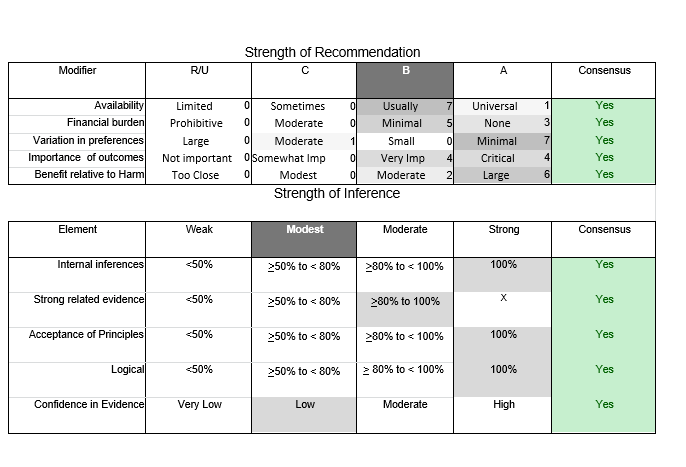
A4. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal dominant inheritance, distal weakness, myotonic discharges on EMG, past or family history of Paget disease, frontotemporal dementia, or motor neuron disease, clinicians should perform genetic testing for mutations in VCP (hIBMPFD) (Level B).
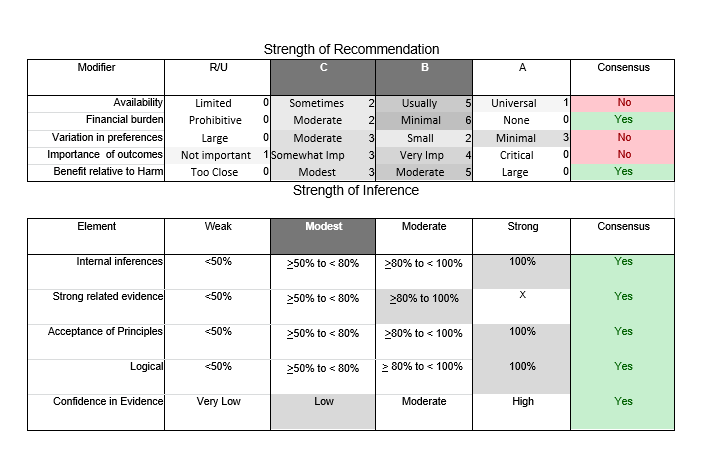
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level B using informal consensus.
A5. In patients with limb-girdle weakness and suspected muscular dystrophy who either do not have clinical features to suggest a specific form of dystrophy or in whom initial genetic testing is not informative, clinicians should perform muscle biopsy in order to delineate characteristic features that direct further genetic testing (such as immunohistochemistry/immunoblotting for various sarcolemmal proteins, calpain-3, or features of myofibrillar myopathy; see figures 4 and 5) or to exclude an alternative diagnosis (e.g., a metabolic myopathy, mitochondrial myopathy, congenital myopathy, or inflammatory myopathy) (Level B).
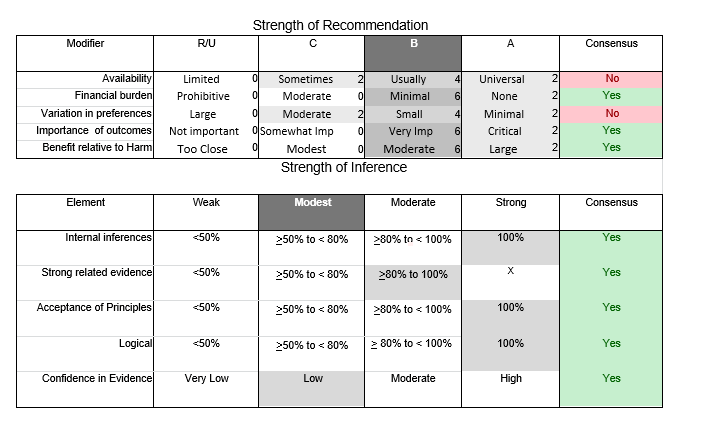
A6. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal recessive inheritance, scapular winging but no calf hypertrophy, and normal cardiorespiratory function, clinicians should perform initial genetic testing for mutations in calpain-3 (LGMD2A). Patients of English, French, Spanish, Italian, Portuguese, or Brazilian descent may have a higher pretest probability of this disorder (Level B).
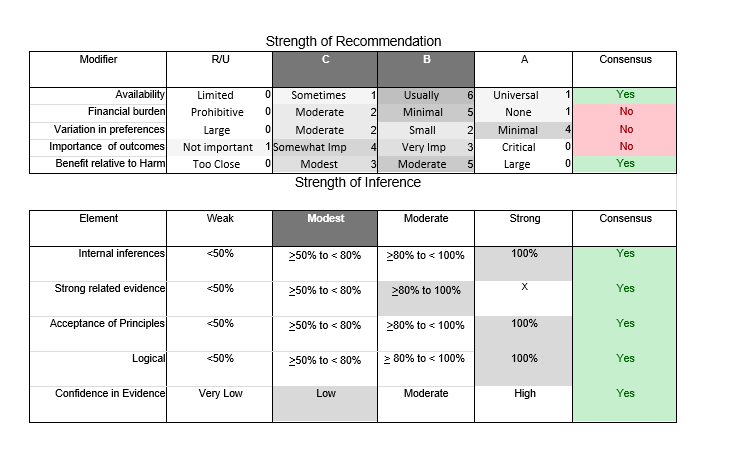
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level B using informal consensus.
A7. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal recessive inheritance and calf atrophy and weakness (i.e., inability to stand on toes), clinicians should perform genetic testing for mutations in anoctamin-5 (LGMD2L) or dysferlin (LGMD2B). If the onset of symptoms is in the teens or early 20s or the patient is from Asia, clinicians should assess for dysferlin mutations first and, if negative, test for anoctamin-5 mutations. If the onset of symptoms is in the 30s or later or the patient is of English or northern European ancestry, clinicians should assess for anoctamin-5 mutations first and, if negative, test for dysferlin mutations (Level B).
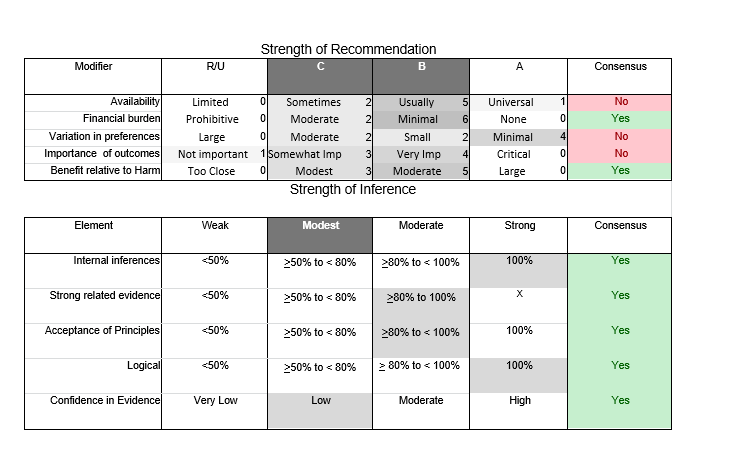
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level B using informal consensus.
A8. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal recessive inheritance and muscle biopsy immunohistochemistry showing reduction in α-, β-, γ-, or δ-sarcoglycans, clinicians should perform genetic testing for mutations in the sarcoglycan genes (LGMD2C–2F) (Level B).
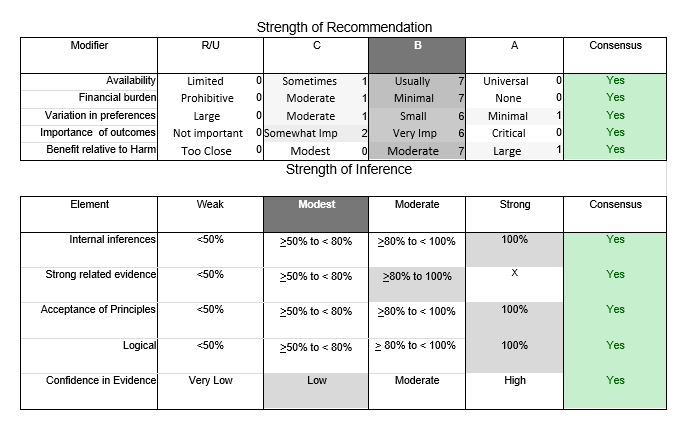
A9. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal recessive inheritance who are of Hutterite descent, clinicians should perform genetic testing for mutations in TRIM32 (LGMD2H) (Level B).
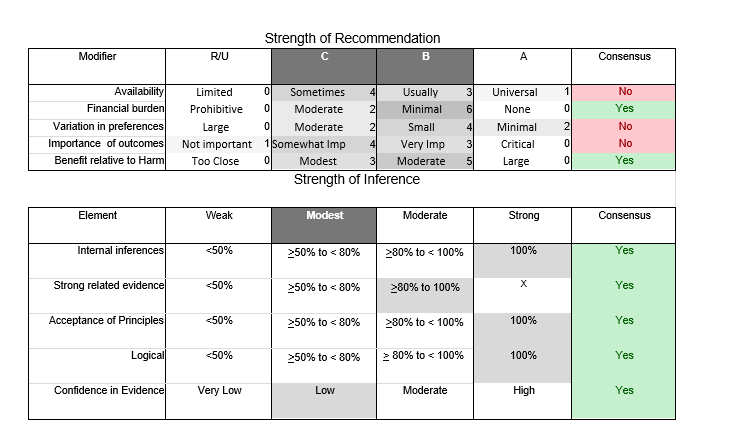
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level B using informal consensus.
A10. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal recessive inheritance, scapular winging, calf hypertrophy, and early cardiorespiratory involvement, clinicians should perform initial genetic testing for mutations in FKRP (LGMD2I). Patients of northern European descent may have a higher pretest probability of this disorder (Level B).
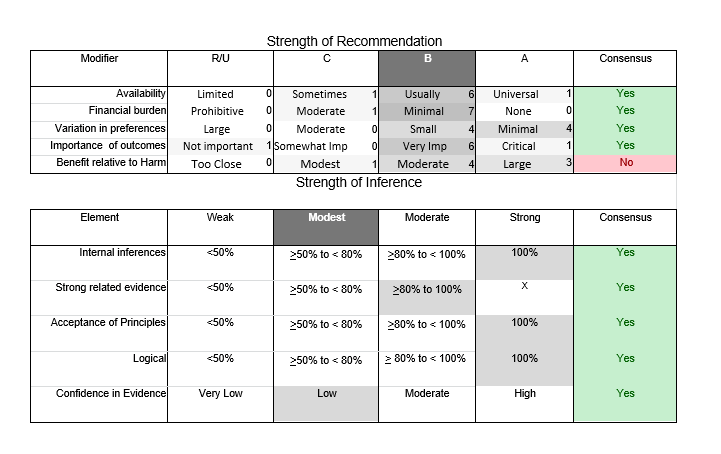
A11. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal recessive inheritance and mental retardation, clinicians should screen for mutations in genes that cause primary or secondary deficiency of α-dystroglycan (LGMD2K, LGMD2M, LGMD2N, LGMD2O, and LGMD2P) (Level B).
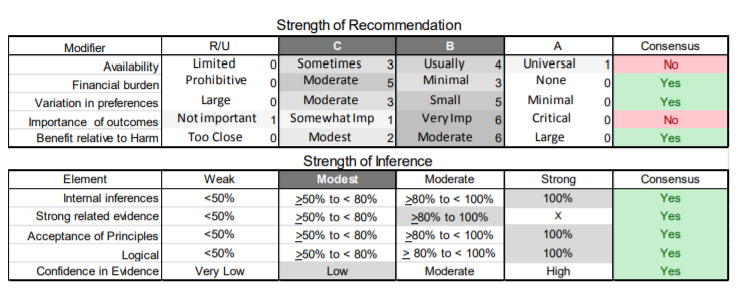
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level B using informal consensus.
A12. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal recessive inheritance and epidermolysis bullosa or pyloric atresia, clinicians should perform genetic testing for mutations in plectin (Level B).
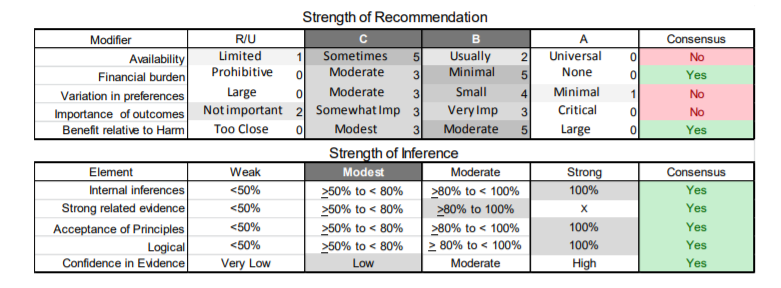
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level B using informal consensus.
A13. In male patients with limb-girdle weakness and suspected muscular dystrophy with probable X-linked inheritance, clinicians should perform genetic testing for mutations in the dystrophin gene (Duchenne or Becker muscular dystrophy) (Level B).
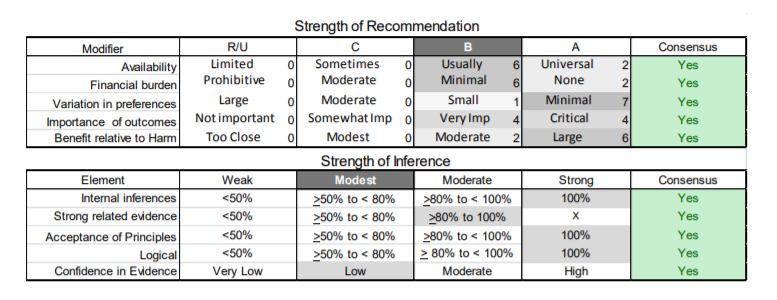
A14. In patients with limb-girdle weakness and suspected muscular dystrophy with probable autosomal recessive inheritance and no other specific clinical features or in whom muscle biopsy does not inform genetic testing, clinicians should perform dried blood spot test for α-glucosidase (acid maltase) deficiency or Pompe disease (Level B).
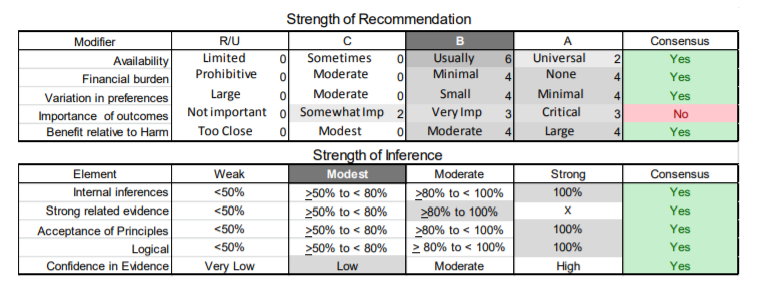
A15. In female patients with limb-girdle weakness and suspected muscular dystrophy with probable X-linked inheritance, clinicians should perform genetic testing for dystrophin mutations or perform a muscle biopsy and immunostain for dystrophin to assess for a mosaic pattern of staining. If abnormal immunostaining is present, clinicians should confirm the diagnosis of manifesting carrier of dystrophinopathy with genetic testing for mutations in the dystrophin gene (Level B).
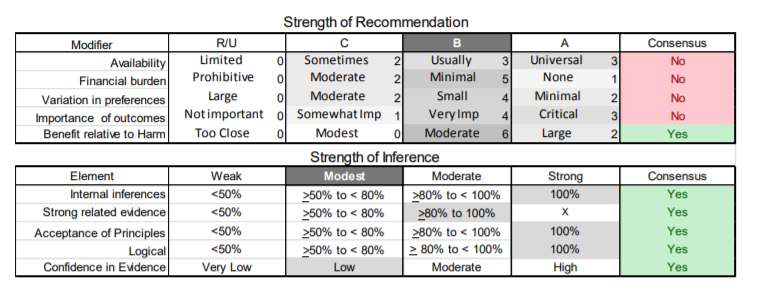
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level B using informal consensus.
Humeroperoneal weakness (figure e-1).
B1. In patients with humeroperoneal weakness and suspected muscular dystrophy with probable autosomal dominant inheritance, early cardiac involvement, and no joint laxity, clinicians should perform genetic testing for mutations in the lamin A/C gene (AD-EDMD, LGMD1B). If the inheritance pattern is probably X-linked, clinicians should perform genetic testing for mutations in the emerin gene (XR-EDMD) (Level B).
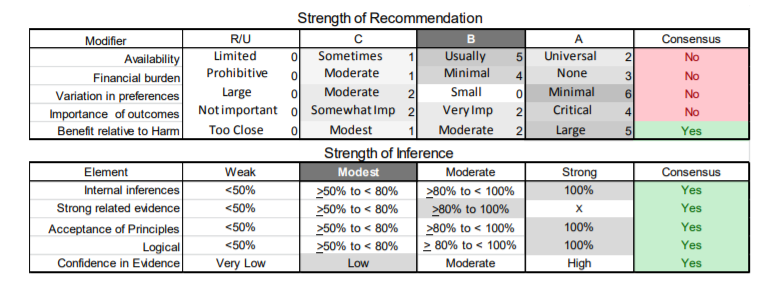
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level B using informal consensus.
B2. In patients with humeroperoneal weakness and suspected muscular dystrophy with early cardiac involvement and no joint laxity who do not possess mutations in the lamin A/C or emerin gene, clinicians should perform muscle biopsy to delineate characteristic abnormalities that direct further genetic testing (see figure e-1 for muscle biopsy features that direct genetic testing) (Level B).
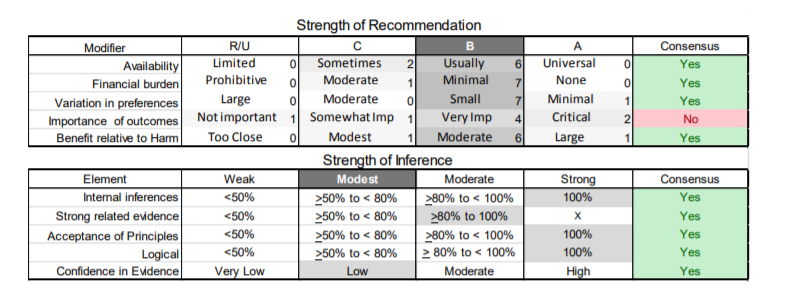
B3. In patients with humeroperoneal weakness and suspected muscular dystrophy with probable autosomal dominant inheritance, joint laxity, protuberant calcaneus, and no cardiac involvement, clinicians should perform genetic testing for mutations in the collagen VI gene (Bethlem myopathy). If the inheritance pattern is probably autosomal recessive with congenital onset, clinicians should perform genetic testing for mutations in the collagen VI gene (Ullrich myopathy) (Level B).
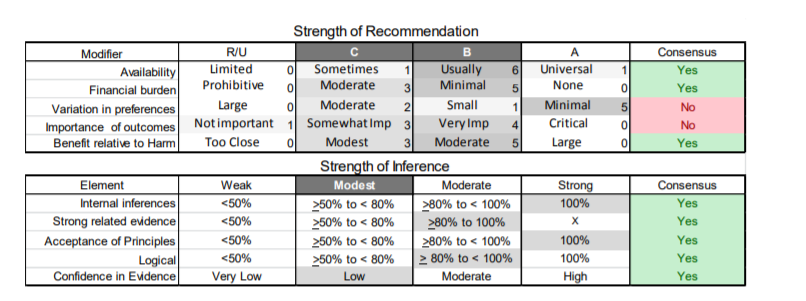
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level B using informal consensus.
Distal muscular dystrophy.
C1. In patients with late adult onset of index finger and wrist extensor weakness followed by atrophy and weakness of hand muscles and muscle biopsy showing rimmed vacuoles, clinicians should make a diagnosis of Welander distal myopathy. Patients of Swedish or Finnish descent may have a higher pretest probability of this disorder. Clinicians should confirm the diagnosis with genetic testing for Welander myopathy when testing becomes commercially available (Level B).
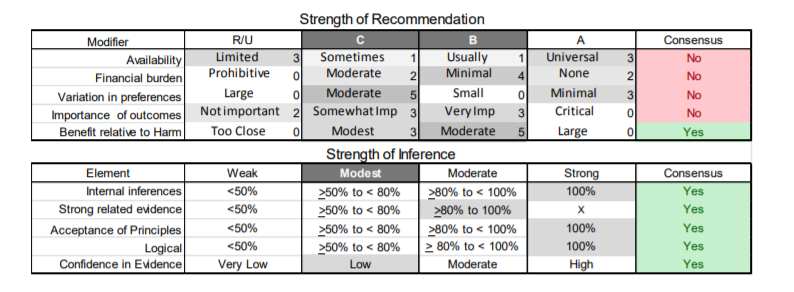
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level B using informal consensus.
C2. In patients with suspected distal muscular dystrophy and probable autosomal recessive inheritance with early onset of calf weakness, clinicians should perform genetic testing for mutations in the anoctamin-5 and dysferlin genes. If the patient is of northern European descent, clinicians should perform initial genetic testing for mutations in the anoctamin-5 gene (LGMD2L) and, if negative, perform genetic testing for mutations in the dysferlin gene (LGMD2B). If the patient is from eastern Asia (Japan, China, Korea), clinicians should perform initial genetic testing for mutations in the dysferlin gene (LGMD2B, Miyoshi myopathy) and, if negative, perform genetic testing for mutations in the anoctamin-5 gene (LGMD2L) (Level B).
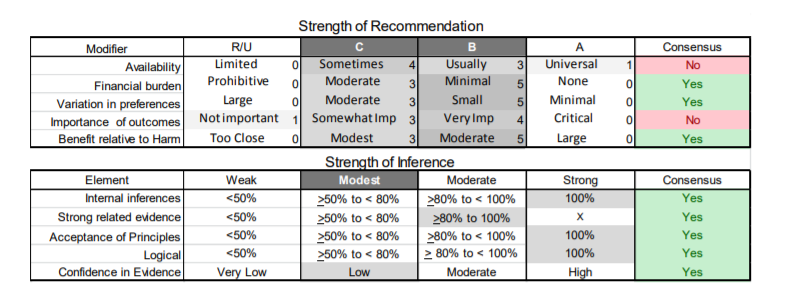
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level B using informal consensus.
C3. In patients with suspected distal muscular dystrophy and probable autosomal recessive inheritance with early onset (<30 years of age) of progressive foot drop who are of Japanese or Middle Eastern Jewish descent, clinicians should perform initial genetic testing for GNE mutations (AR-hIBM) (Level B).
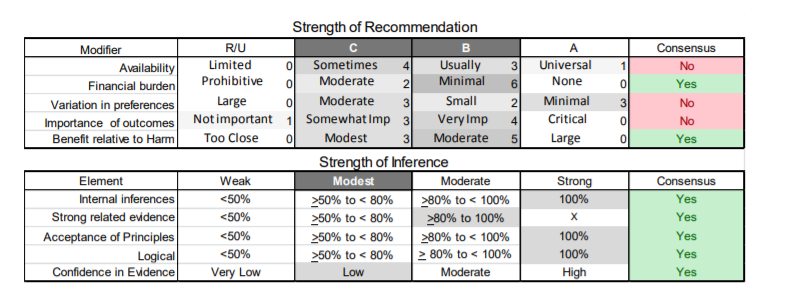
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level B using informal consensus.
C4. In patients with suspected distal muscular dystrophy without the clinical features in C2 or C3 above, clinicians should perform a muscle biopsy to direct further genetic testing (see figure e-2 for biopsy and clinical features that direct genetic testing) (Level B).
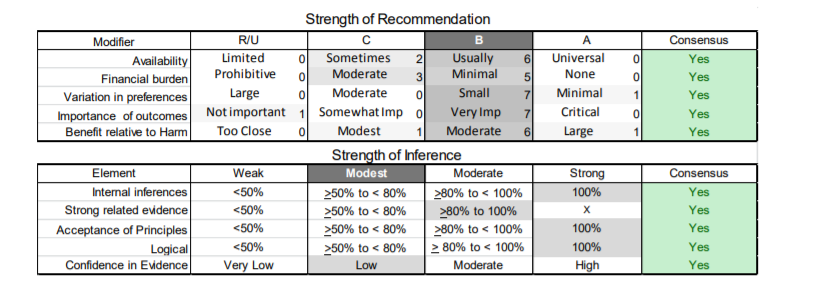
Other diagnostic considerations.
D1. In patients with muscular dystrophy who have proximal as well as distal weakness, clinicians should use specific clinical features (e.g., rippling muscles, cardiomyopathy, atrophy of specific muscle groups, irritability on EMG) and biopsy features (MFM, reduction of emerin immunostaining, presence of rimmed vacuoles) to guide genetic testing, which may include mutations in the genes causing the various forms of MFM (see section on MFM), LGMD2B (dysferlin), LGMD2L (anoctamin-5), LGMD2J (titin), LGMD1C (caveolin-3), and EDMD (emerin and lamin A/C) (Level B).
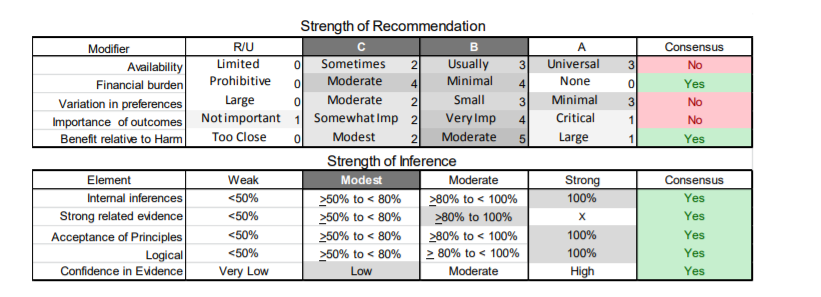
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level B using informal consensus.
D2. In patients with suspected muscular dystrophy in whom initial genetic testing, muscle biopsy, and dried blood spot test for Pompe disease do not provide a diagnosis, clinicians may obtain genetic consultation or perform parallel sequencing of targeted exomes, whole-exome sequencing, whole-genome screening, or next-generation sequencing to identify the genetic abnormality (Level C).
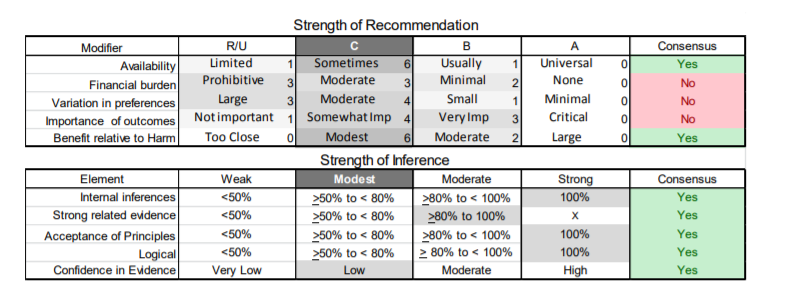
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level C using informal consensus.
Evaluation and Medical Management of Muscular Dystrophies
Cardiac involvement in muscular dystrophies.
E1. Clinicians should refer newly diagnosed patients with
- LGMD1A, LGMD1B, LGMD1D, LGMD1E, LGMD2C–K, LGMD2M–P, BMD, EDMD, and MFM
- muscular dystrophy without a specific genetic diagnosis for cardiology evaluation, including ECG and structural evaluation (echocardiography or cardiac MRI), even if they are asymptomatic from a cardiac standpoint, to guide appropriate management (Level B).
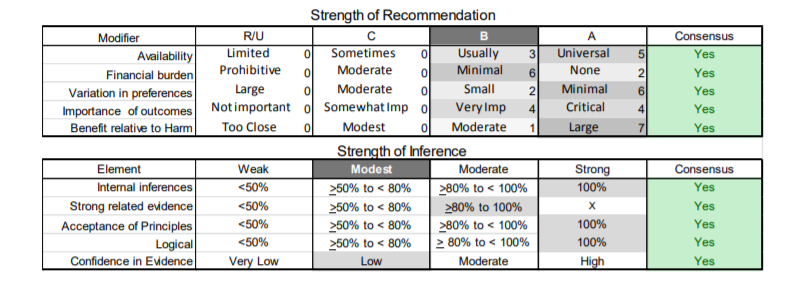
E2. Clinicians should refer muscular dystrophy patients with palpitations or who are found to have symptomatic or asymptomatic tachycardia or arrhythmias for cardiology evaluation (Level B).
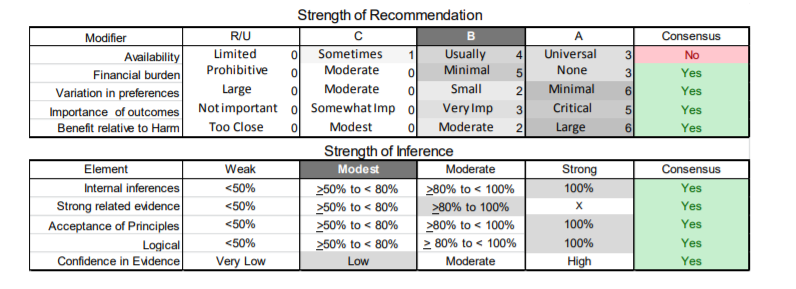
E3. Clinicians should refer muscular dystrophy patients with signs or symptoms of cardiac failure for cardiology evaluation (e.g., medical management, left ventricular assist device placement, or cardiac transplantation, as deemed necessary by the cardiologist) to prevent cardiac death (Level B).
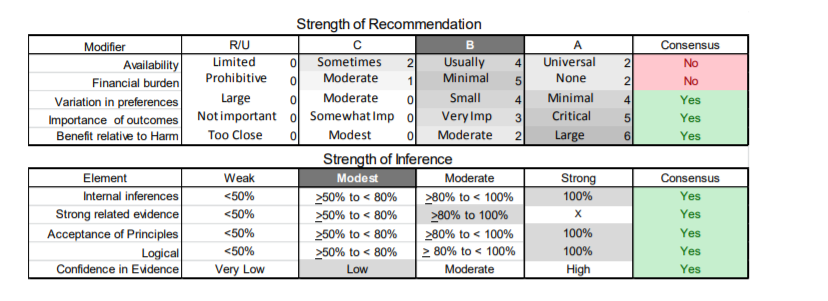
E4. It is not obligatory for clinicians to refer patients with LGMD2A, LGMD2B, and LGMD2L for cardiac evaluation unless they develop overt cardiac signs or symptoms (Level B).
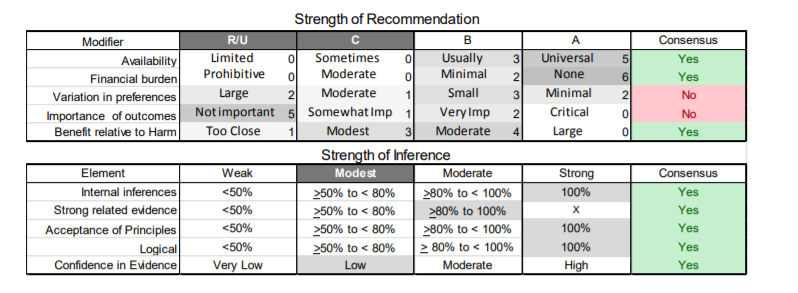
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level B using informal consensus.
E5. Clinicians should encourage female carriers of dystrophinopathy and emerinopathy to seek evaluation by a neuromuscular specialist and a cardiologist to assess for skeletal muscle and cardiac muscle involvement and to proactively treat cardiac involvement (Level B).
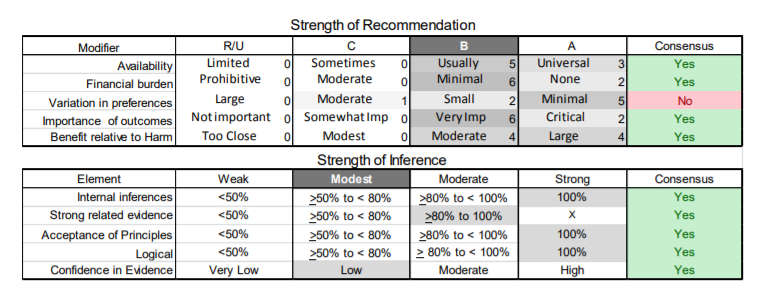
Dysphagia and nutrition.
F1. Clinicians should refer muscular dystrophy patients with dysphagia, frequent aspiration, or weight loss for swallowing evaluation and/or gastroenterology evaluation to assess and manage swallowing function and aspiration risk, to teach patients techniques for safe and effective swallowing (e.g., “chin tuck” maneuver, altered food consistencies, etc.), and to consider placement of a gastrostomy/jejunostomy tube for nutritional support (Level B).
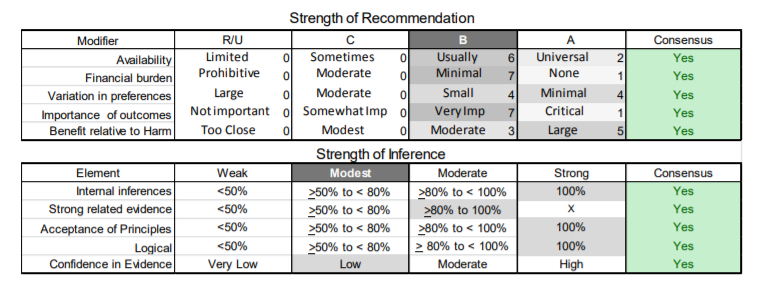
Pulmonary complications.
G1. Clinicians should order pulmonary function testing (spirometry and maximal inspiratory/expiratory force in the upright and, if normal, supine positions) or refer for pulmonary evaluation (to identify and treat respiratory insufficiency) in muscular dystrophy patients at the time of diagnosis, or if they develop pulmonary symptoms later in their course (Level B).
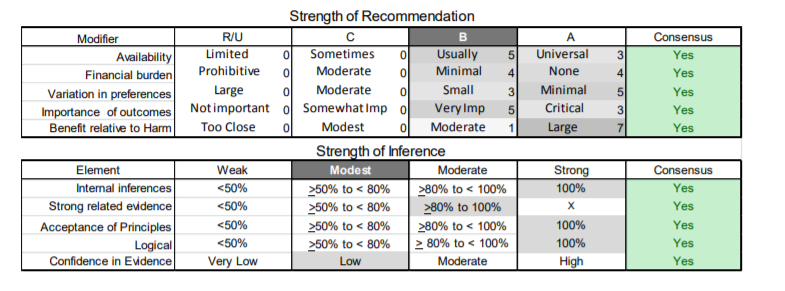
G1a. In patients with a known high risk of respiratory failure (e.g., those with LGMD2I or MFM), clinicians should obtain periodic pulmonary function testing (spirometry and maximal inspiratory/expiratory force in the upright position and, if normal, in the supine position) or evaluation by a pulmonologist to identify and treat respiratory insufficiency (Level B).
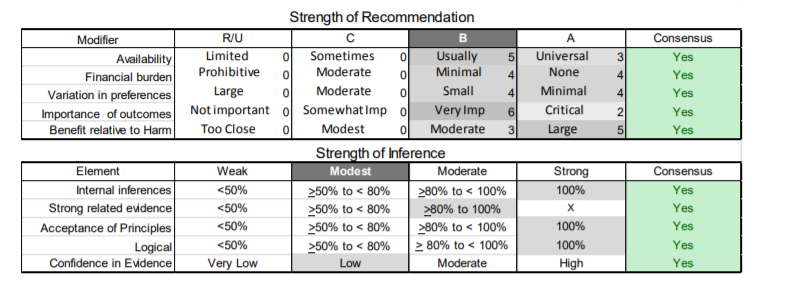
G2. It is not obligatory for clinicians to refer patients with LGMD2B and LGMD2L for pulmonary evaluation or pulmonary function testing unless they are symptomatic (Level C).
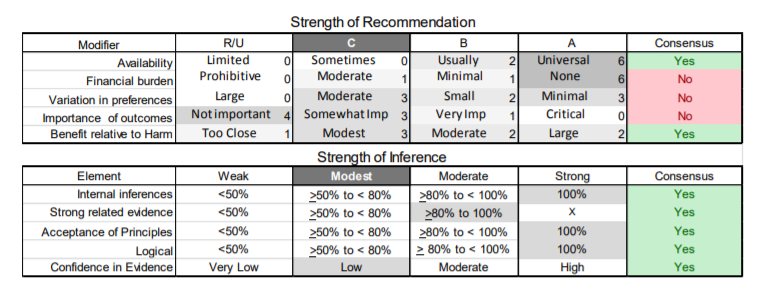
G3. Clinicians should refer muscular dystrophy patients with excessive daytime somnolence, nonrestorative sleep (e.g., frequent nocturnal arousals, morning headaches, excessive daytime fatigue), or respiratory insufficiency based on pulmonary function tests for pulmonary or sleep medicine consultation for consideration of noninvasive ventilation to improve quality of life (Level B).
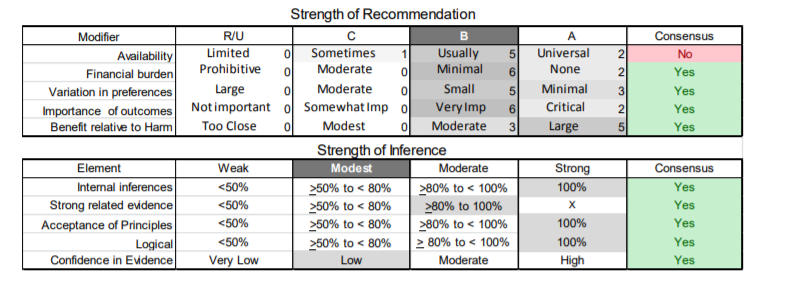
Cognitive dysfunction and learning disabilities.
H1. In muscular dystrophy patients with symptoms suggestive of cognitive dysfunction or learning disabilities, clinicians may order neuropsychological testing, MRI of the brain, and/or developmental pediatrics consultation to assess for and optimally manage CNS involvement (Level C).
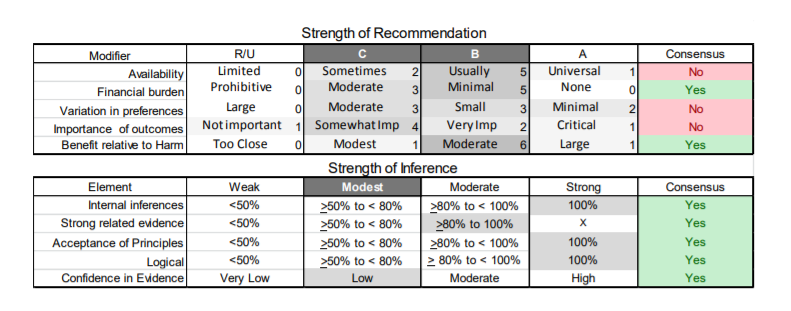
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level C using informal consensus.
Spinal deformities.
I1. Clinicians should monitor patients with muscular dystrophy for the development of spinal deformities to prevent resultant complications and preserve function (Level B).
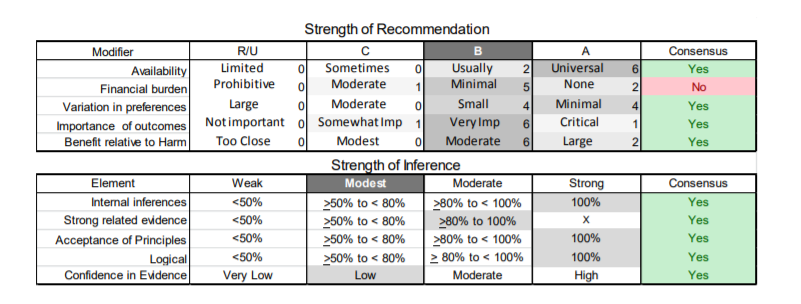
I2. Clinicians should refer muscular dystrophy patients with musculoskeletal spine deformities to an orthopedic spine surgeon for monitoring and surgical intervention if it is deemed necessary in order to maintain normal posture, assist mobility, maintain cardiopulmonary function, and optimize quality of life (Level B).
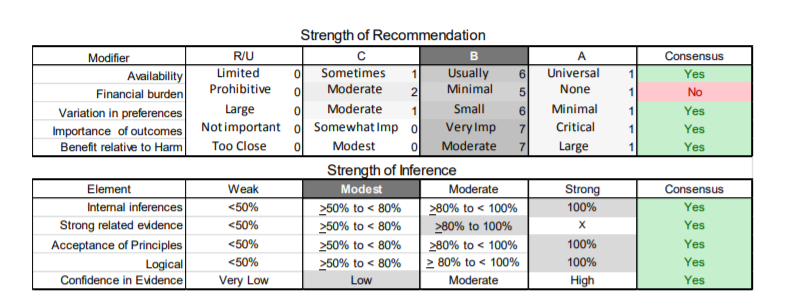
Osteoporosis.
J1. Clinicians may choose to evaluate patients with restricted mobility due to muscular dystrophy with bone density studies for osteoporosis in order to institute timely management and minimize fractures (Level C).
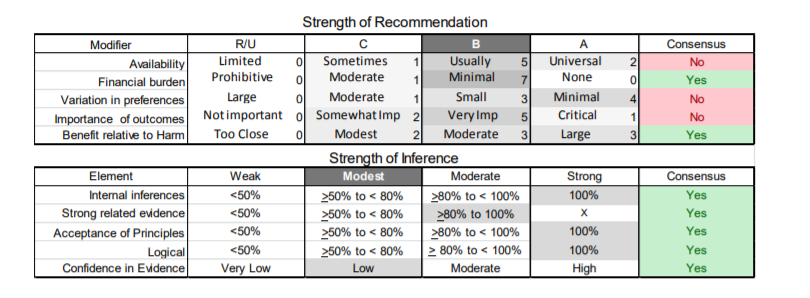
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level C using informal consensus.
Infection prophylaxis.
K1. Clinicians should recommend pneumococcal polysaccharide vaccine (PPSV23) as per the CDC schedulee514 and annual influenza vaccine to patients with muscular dystrophy in order to prevent respiratory complications of pneumococcal pneumonia and influenza (Level B).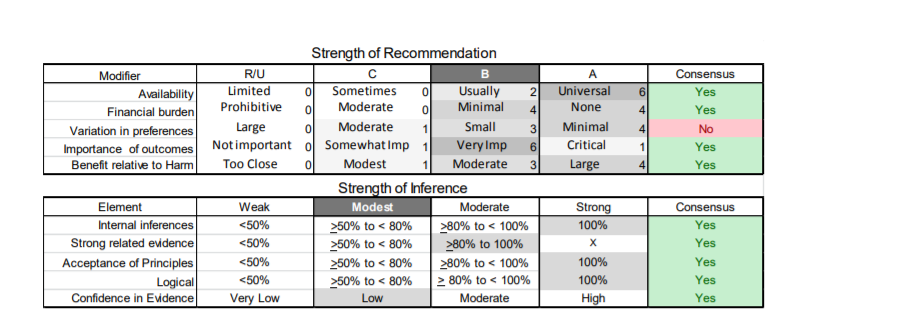
Rehabilitative Management and Treatment of Muscular Dystrophies Clinical rehabilitative management.
L1. Clinicians should refer patients with muscular dystrophy to a clinic that has access to multiple specialties (e.g., physical therapy, occupational therapy, respiratory therapy, speech and swallowing therapy, cardiology, pulmonology, orthopedics, and genetics) designed specifically to care for patients with muscular dystrophy and other neuromuscular disorders in order to provide efficient and effective long-term care (Level B).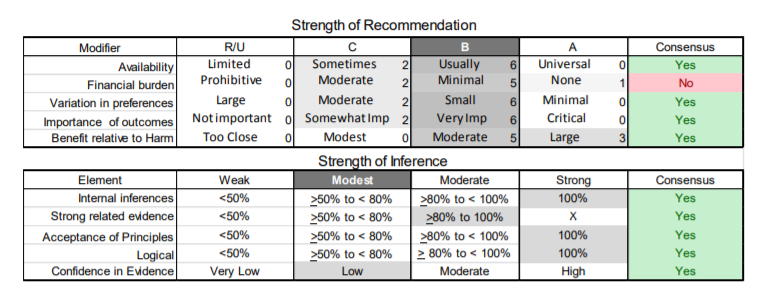
L2. Clinicians might discuss opportunities for participation in clinical trials, if available, with muscular dystrophy patients (Level C).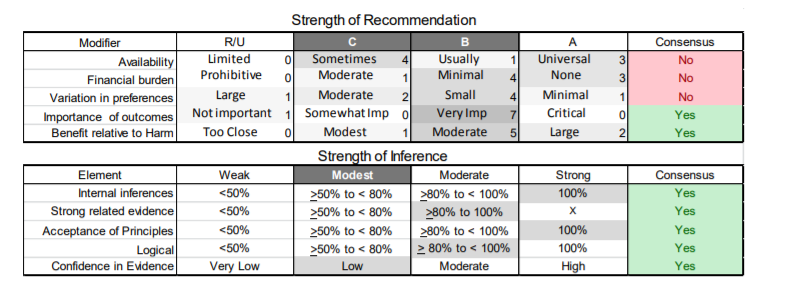 There is a persistent, substantial lack of consensus regarding availability. The recommendation defaults to Level C.
There is a persistent, substantial lack of consensus regarding availability. The recommendation defaults to Level C.
L3. Clinicians should recommend that patients with muscular dystrophy have periodic assessments by a physical and occupational therapist for symptomatic and preventive screening (Level B).
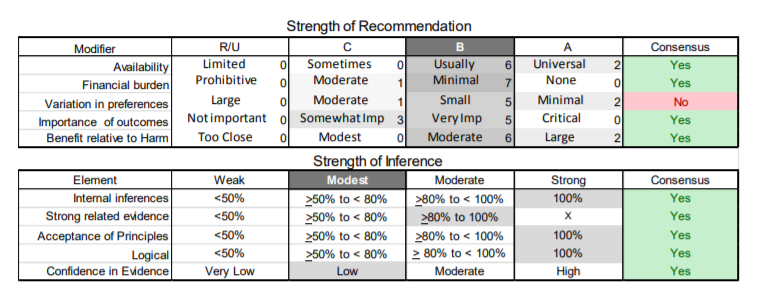
L4. While respecting and protecting patient autonomy, clinicians should proactively anticipate and facilitate patient and family decision making as the disease progresses, including decisions regarding loss of mobility, need for assistance with activities of daily living, medical complications, and end-of-life care (Level B).
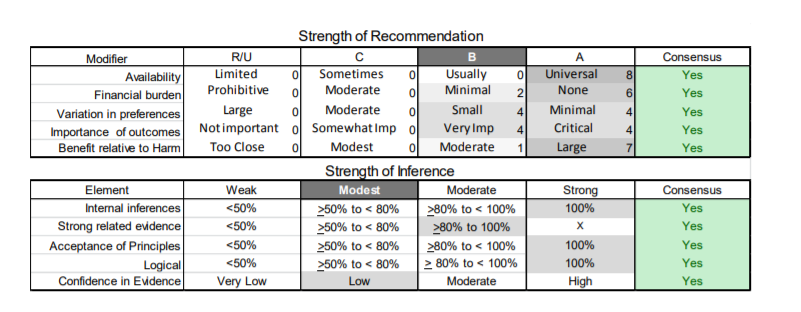
L5. For patients with muscular dystrophy, clinicians should prescribe physical and occupational therapy, as well as bracing and assistive devices that are adapted specifically to the patient’s deficiencies and contractures, in order to preserve mobility and function and prevent contractures (Level B).
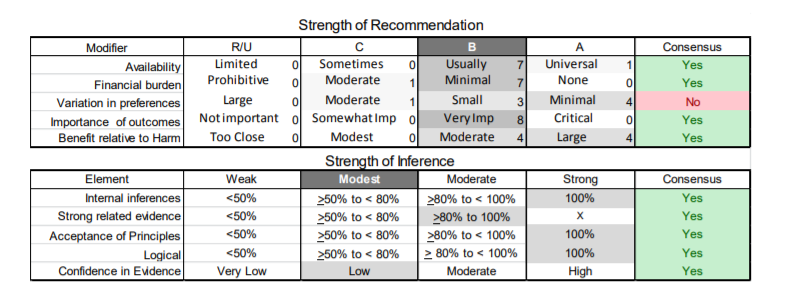
Strength training and aerobic exercise training.
M1. Clinicians may advise patients with muscular dystrophy that aerobic exercise combined with a supervised submaximal strength training program is probably safe (Level C).
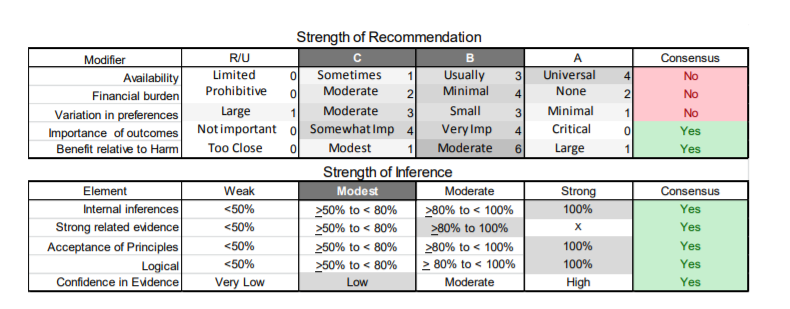
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level C using informal consensus.
M2. Clinicians may advise patients with muscular dystrophy that gentle, low-impact aerobic exercise (swimming, stationary bicycling) improves cardiovascular performance, increases muscle efficiency, and lessens fatigue (Level C).
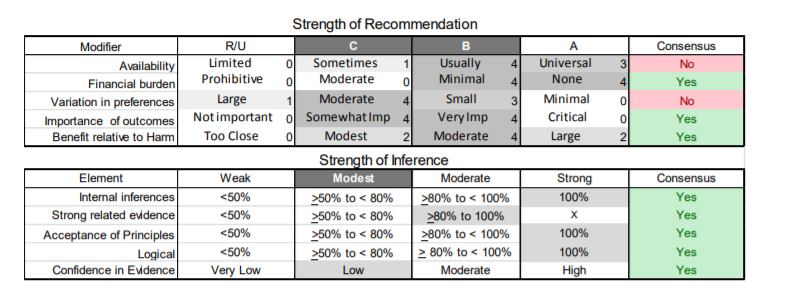
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level C using informal consensus.
M3. Clinicians may counsel patients with muscular dystrophy to hydrate adequately, not to exercise to exhaustion, and to avoid supramaximal, high-intensity exercise (Level C).
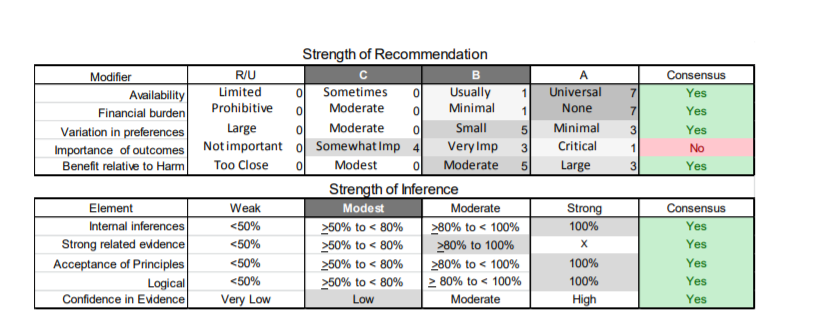
There was substantial consensus after round 2. Based on the modifiers (availability, financial burden, variations), the panel selected Level C using informal consensus.
M4. Clinicians should educate patients with muscular dystrophy who are participating in an exercise program about the warning signs of overwork weakness and myoglobinuria, which include feeling weaker rather than stronger within 30 minutes after exercise, excessive muscle soreness 24–48 hours following exercise, severe muscle cramping, heaviness in the extremities, and prolonged shortness of breath (Level B).
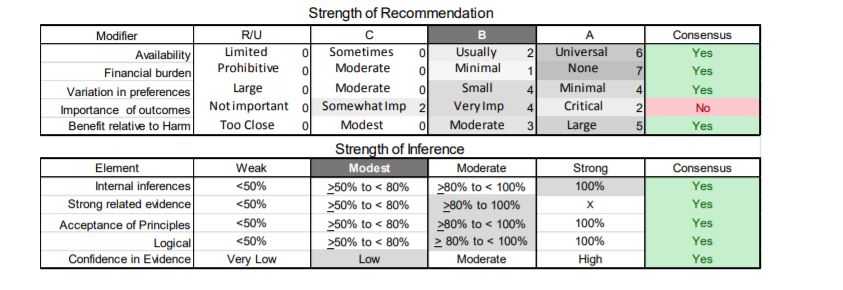
Medical treatments.
N1. Clinicians should not offer patients with LGMD gene therapy outside of a research study designed to determine the efficacy and safety of the treatment (Level R).
N2. Clinicians should not offer patients with LGMD neutralizing antibody to myostatin outside of a research study designed to determine the efficacy and safety of the treatment (Level R).
N3. Clinicians should not offer patients with BMD myoblast transplantation or subcutaneous growth hormone injections outside of a research study designed to determine the efficacy and safety of the treatment (Level R).
Level R determinations based on insufficient evidence regarding efficacy and judgment of high cost or risk of interventions.
References
e1. Walton, JN, Nattrass FJ. On the classification, natural history and treatment of the myopathies. Brain 1954;77(2):169–231.
e2. Bushby KM. Diagnostic criteria for the limb-girdle muscular dystrophies: report of the
ENMC Consortium on Limb-Girdle Dystrophies. Neuromuscul Disord 1995;5(1):71–74.
e3. Bushby KM, Beckmann JS. The limb-girdle muscular dystrophies--proposal for a new nomenclature. Neuromuscul Disord 1995;5(4):337–343.
e4. Guglieri M, Straub V, Bushby K, Lochmüller H. Limb-girdle muscular dystrophies. Curr
Opin Neurol 2008;21(5):576–584.
e5. Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain 2009;132(Pt 11):3175–3186.
e6. Bushby K. Diagnosis and management of the limb girdle muscular dystrophies. Pract Neurol
2009;9(6):314–323.
e7. AAN (American Academy of Neurology). 2004. Clinical Practice Guideline Process Manual,
2004 Ed. St. Paul, MN: The American Academy of Neurology.
e8. Hauser MA, Conde CB, Kowaljow V, et al. myotilin Mutation found in second pedigree with
LGMD1A. Am J Hum Genet 2002;71(6):1428–1432.
e9. Selcen D, Engel AG. Mutations in myotilin cause myofibrillar myopathy. Neurology
2004;62(8):1363–1371.
e10. Moore SA, Shilling CJ, Westra S, et al. Limb-girdle muscular dystrophy in the United
States. J Neuropathol Exp Neurol 2006;65(10):995–1003.
e11. Lo HP, Cooper ST, Evesson FJ, et al. Limb-girdle muscular dystrophy: diagnostic evaluation, frequency and clues to pathogenesis. Neuromuscul Disord 2008;18(1):34–44.
e12. Benedetti S, Menditto I, Degano M, et al. Phenotypic clustering of lamin A/C mutations in neuromuscular patients. Neurology 2007;69:1285–1292.
e13. Fanin M, Nascimbeni AC, Aurino S, et al. Frequency of LGMD gene mutations in Italian patients with distinct clinical phenotypes. Neurology 2009;72(16):1432–1435.
e14. Garg A, Speckman RA, Bowcock AM. Multisystem dystrophy syndrome due to novel missense mutations in the amino-terminal head and alpha-helical rod domains of the lamin A/C gene. Am J Med 2002;112:549–555.
e15. van Tintelen JP, Hofstra RM, Katerberg H, et al; Working Group on Inherited Cardiac Disorders, line 27/50, Interuniversity Cardiology Institute of The Netherlands. High yield of LMNA mutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics. Am Heart J 2007;154:1130–1139.
e16. Vytopil M, Benedetti S, Ricci E, et al. Mutation analysis of the lamin A/C gene (LMNA) among patients with different cardiomuscular phenotypes. J Med Genet 2003;40:e132.
e17. Guglieri M, Magri F, D’Angelo MG, et al. Clinical, molecular, and protein correlations in a large sample of genetically diagnosed Italian limb girdle muscular dystrophy patients. Hum Mutat 2008;29:258–266.
e18. Fanin M, Nascimbeni AC, Fulizio L, Angelini C. The frequency of limb girdle muscular dystrophy 2A in northeastern Italy. Neuromuscular Disord 2005;15:218-224.
e19. Passos-Bueno MR, Moreira ES, Marie SK, et al. Main clinical features of the three mapped autosomal recessive limb-girdle muscular dystrophies and estimated proportion of each form in 13 Brazilian families. J Med Genet 1996;33(2):97–102.
e20. Dinçer P, Leturcq F, Richard I, et al. A biochemical, genetic, and clinical survey of autosomal recessive limb girdle muscular dystrophies in Turkey. Ann Neurol 1997;42(2):222–229.
e21. Richard I, Brenguier L, Dinçer P, et al. Multiple independent molecular etiology for limbgirdle muscular dystrophy type 2A patients from various geographical origins. Am J Hum Genet 1997;60(5):1128–1138.
e22. Chou FL, Angelini C, Daentl D, et al. Calpain III mutation analysis of a heterogeneous limb-girdle muscular dystrophy population. Neurology 1999;52(5):1015–1020.
e23. Passos-Bueno MR, Vainzof M, Moreira ES, Zatz M. Seven autosomal recessive limb-girdle muscular dystrophies in the Brazilian population: from LGMD2A to LGMD2G. Am J Med Genet 1999;82(5):392–398.
e24. Dinçer P, Akçören Z, Demir E, et al. A cross section of autosomal recessive limb-girdle muscular dystrophies in 38 families. J Med Genet 2000;37(5):361–367.
e25. Chae J, Minami N, Jin Y, et al. Calpain 3 gene mutations: genetic and clinico-pathologic findings in limb-girdle muscular dystrophy. Neuromuscul Disord 2001;11(6-7):547–555.
e26. Nonaka I, Minami N, Chae J, et al. Limb-girdle muscular dystrophy research in Japan. Acta
Myol 2001;20:83–86.
e27. Piluso G, Politano L, Aurino S, et al. Extensive scanning of the calpain-3 gene broadens the spectrum of LGMD2A phenotypes. J Med Genet 2005;42(9):686–693.
e28. Sáenz A, Leturcq F, Cobo AM, et al. LGMD2A: genotype-phenotype correlations based on a large mutational survey on the calpain 3 gene. Brain 2005;128(Pt 4):732–742.
e29. Krahn M, Bernard R, Pecheux C, et al; Calpain Study Group of the French LGMD Network. Screening of the CAPN3 gene in patients with possible LGMD2A. Clin Genet 2006;69(5):444–449.
e30. Hanisch F, Müller CR, Grimm D, et al. Frequency of calpain-3 c.550delA mutation in limb girdle muscular dystrophy type 2 and isolated hyperCKemia in German patients. Clin Neuropathol 2007;26(4):157–163.
e31. Shin JH, Kim HS, Lee CH, Kim CM, Park KH, Kim DS. Mutations of CAPN3 in Korean patients with limb-girdle muscular dystrophy. J Korean Med Sci 2007;22(3):463–469.
e32. Duno M, Sveen ML, Schwartz M, Vissing J. cDNA analyses of CAPN3 enhance mutation detection and reveal a low prevalence of LGMD2A patients in Denmark. Eur J Hum Genet 2008;16(8):935–940.
e33. Todorova A, Georgieva B, Tournev I, et al. A large deletion and novel point mutations in the calpain 3 gene (CAPN3) in Bulgarian LGMD2A patients. Neurogenetics 2007;8:225229.
e34. van der Kooi AJ, Frankhuizen WS, Barth PG, et al. Limb-girdle muscular dystrophy in the
Netherlands: gene defect identified in half the families. Neurology 2007;68:2125–2128.
e35. Krahn M, Béroud C, Labelle V, et al. Analysis of the DYSF mutational spectrum in a large cohort of patients. Hum Mutat 2009;30:E345–E75.
e36. Tagawa K, Ogawa M, Kawabe K, et al. Protein and gene analyses of dysferlinopathy in a large group of Japanese muscular dystrophy patients. J Neurol Sci 2003;211:23–28.
e37. Fanin M, Duggan DJ, Mostacciuolo ML, et al. Genetic epidemiology of muscular dystrophies resulting from sarcoglycan gene mutations. J Med Genet 1997;34:973–977.
e38. Duggan DJ, Gorospe JR, Fanin M, Hoffman EP, Angelini C. Mutations in the sarcoglycan genes in patients with myopathy. N Eng J Med 1997;336:618–624.
e39. Ginjaar HB, van der Kooi AJ, Ceelie H, et al. Sarcoglycanopathies in Dutch patients with autosomal recessive limb girdle muscular dystrophy. J Neurol 2000;247:524–529.
e40. Khadilkar SV, Singh RK, Hegde M, Urtizberea A, Love DR, Chong B. Spectrum of mutations in sarcoglycan genes in the Mumbai region of western India: high prevalence of 525del T. Neurol India 2009;57(4):406–410.
e41. McNally EM, Duggan D, Gorospe JR, et al. Mutations that disrupt the carboxyl-terminus of gamma-sarcoglycan cause muscular dystrophy. Hum Mol Genet 1996;5:1841–1847.
e42. McNally EM, Passos-Bueno MR, Bönnemann CG, et al. Mild and severe muscular dystrophy caused by a single gamma-sarcoglycan mutation. Am J Hum Genet 1996;59:1040–1047.
e43. Politano L, Nigro V, Passamano L, et al. Evaluation of cardiac and respiratory involvement in sarcoglycanopathies. Neuromuscul Disord 2001;11(2):178–185.
e44. Moreira ES, Vainzof M, Suzuki OT, Pavanello RC, Zatz M, Passos-Bueno MR. Genotypephenotype correlations in 35 Brazilian families with sarcoglycanopathies including the description of three novel mutations. J Med Genet 2003;40:E12. e45. Angelini C, Fanin M, Freda MP, Duggan DJ, Siciliano G, Hoffman EP. The clinical spectrum of sarcoglycanopathies. Neurology 1999;52:176–179.
e46. Boyden SE, Salih MA, Duncan AR, et al. Efficient identification of novel mutations in patients with limb girdle muscular dystrophy. Neurogenetics 2010;11:449–455.
e47. Duggan DJ, Fanin M, Pegoraro E, Angelini C, Hoffman EP. alpha-Sarcoglycan (adhalin) deficiency: complete deficiency patients are 5% of childhood-onset dystrophin-normal muscular dystrophy and most partial deficiency patients do not have gene mutations. J
Neurol Sci 1996;140:30–39.
e48. Ljunggren A, Duggan D, McNally E, et al. Primary adhalin deficiency as a cause of muscular dystrophy in patients with normal dystrophin. Ann Neurol 1995;38:367–372.
e49. Moreira ES, Vainzof M, Marie SK, Nigro V, Zatz M, Passos-Bueno MR. A first missense mutation in the delta sarcoglycan gene associated with a severe phenotype and frequency of limb-girdle muscular dystrophy type 2F (LGMD2F) in Brazilian sarcoglycanopathies. J Med Genet 1998;35:951–953.
e50. Duggan DJ, Manchester D, Stears KP, Mathews DJ, Hart C, Hoffman EP. Mutations in the delta-sarcoglycan gene are a rare cause of autosomal recessive limb-girdle muscular dystrophy. Neurogenetics 1997;1:49–58.
e51. Selcen D, Ohno K, Engel AG. Myofibrillar myopathy: clinical, morphological and genetic studies in 63 patients. Brain 2004;127(Pt 2):439–451.
e52. Hanisch F, Grimm D, Zierz S, Deschauer M. Frequency of the FKRP mutation c.826C>A in isolated hyperCKemia and in limb girdle muscular dystrophy type 2 in German patients. J Neurol 2010;257:300–301.
e53. Boito CA, Melacini P, Vianello A, et al. Clinical and molecular characterization of patients with limb-girdle muscular dystrophy type 2I. Arch Neurol 2005;62:1894–1899.
e54. Brockington M, Yuva Y, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet 2001;10:2851–2859.
e55. Schwartz M, Hertz JM, Sveen ML, Vissing J. LGMD2I presenting with a characteristic
Duchenne or Becker muscular dystrophy phenotype. Neurology 2005;64:1635–1637.
e56. Kang PB, Feener CA, Estrella E, et al. LGMD2I in a North American population. BMC
Musculoskelet Disord 2007;8:115.
e57. Stensland E, Lindal S, Jonsrud C, et al. Prevalence, mutation spectrum and phenotypic variability in Norwegian patients with Limb Girdle Muscular Dystrophy 2I. Neuromuscul Disord 2011;21:41–46.
e58. Hackman P, Marchand S, Sarparanta J, et al. Truncating mutations in C-terminal titin may cause more severe tibial muscular dystrophy (TMD). Neuromuscul Disord 2008;18:922– 928.
e59. Godfrey C, Clement E, Mein R, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007;130(Pt 10):2725– 2735.
e60. Hicks D, Sarkozy A, Muelas N, et al. A founder mutation in Anoctamin 5 is a major cause of limb-girdle muscular dystrophy. Brain 2011;134:171–182.
e61. Penttilä S, Palmio J, Suominen T, et al. Eight new mutations and the expanding phenotype variability in muscular dystrophy caused by ANO5. Neurology 2012;78:897–903.
e62. Bushby KM, Thambyayah M, Gardner-Medwin D. Prevalence and incidence of Becker muscular dystrophy. Lancet 1991;337:1022-1024.
e63. Siciliano G, Tessa A, Renna M, Manca ML, Mancuso M, Murri L. Epidemiology of dystrophinopathies in North-West Tuscany: a molecular genetics-based revisitation. Clin
Genet 1999;56:51–58.
e64. Nakagawa M, Nakahara K, Yoshidome H, et al. Epidemiology of progressive muscular dystrophy in Okinawa, Japan. Classification with molecular biological techniques.
Neuroepidemiology 1991;10:185–191.
e65. Arikawa E, Hoffman EP, Kaido M, Nonaka I, Sugita H, Arahata K. The frequency of patients with dystrophin abnormalities in a limb-girdle patient population. Neurology 1991;41:1491–1496.
e66. Angelini C, Fanin M, Freda MP, et al. Prognostic factors in mild dystrophinopathies. J
Neurol Sci 1996;142:70–78.
e67. Doriguzzi C, Palmucci L, Mongini T, Chiadò-Piat L, Maniscalco M, Restagno G. Systematic use of dystrophin testing in muscle biopsies: results in 201 cases. Eur J Clin Invest 1997;27:352–358.
e68. Hoffman EP, Kunkel LM, Angelini C, Clarke A, Johnson M, Harris JB. Improved diagnosis of Becker muscular dystrophy by dystrophin testing. Neurology 1989;39:1011–1017.
e69. Sveen ML, Schwartz M, Vissing J. High prevalence and phenotype-genotype correlations of limb girdle muscular dystrophy type 2I in Denmark. Ann Neurol 2006;59:808–815.
e70. Legum C, Shomrat R, Glassner M, Shiloh Y. A molecular survey of Israeli Duchenne and
Becker muscular dystrophy patients. Biomed Pharmacother 1994;48:359–364.
e71. Liang WC, Mitsuhashi H, Keduka E, et al. TMEM43 mutations in Emery-Dreifuss muscular dystrophy-related myopathy. Ann Neurol 2011;69:1005–1013.
e72. Selcen D, Engel AG. Mutations in ZASP define a novel form of muscular dystrophy in humans. Ann Neurol 2005;57:269–276.
e73. Selcen D, Muntoni F, Burton BK, et al. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol 2009;65:83–89.
e74. Kaback M, Lopatequi J, Portuges AR, et al. Genetic screening in the Persian Jewish community: A pilot study. Genet Med 2010;12:628–633.
e75. Bonne G, Mercuri E, Muchir A, et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene.
Ann Neurol 2000;48:170–180.
e76. Boriani G, Gallina M, Merlini L, et al. Clinical relevance of atrial fibrillation/flutter, stroke, pacemaker implant, and heart failure in Emery-Dreifuss muscular dystrophy: a long-term longitudinal study. Stroke 2003;34:901–908.
e77. Carboni N, Mura M, Marrosu G, et al. Muscle imaging analogies in a cohort of patients with different clinical phenotypes caused by LMNA gene mutations. Muscle Nerve
2010;41:458–463.
e78. Charniot J, Desnos M, Zerhouni K, et al. Severe dilated cardiomyopathy and quadriceps myopathy due to lamin A/C gene mutation: a phenotypic study. Eur J Heart Fail
2006;8:249–256.
e79. Chrestian N, Valdmanis PN, Echahidi N, et al. A novel mutation in a large French-Canadian family with LGMD1B. Can J Neurol Sci 2008;35:331–334.
e80. Deconinck N, Dion E, Ben Yaou R, et al. Differentiating Emery-Dreifuss muscular dystrophy and collagen VI-related myopathies using a specific CT scanner pattern.
Neuromuscul Disord 2010;20:517–523.
e81. Hausmanowa-Petrusewicz I, Madej-Pilarczyk A, Marchel M, Opolski G. Emery-Dreifuss dystrophy: a 4-year follow-up on a laminopathy of special interest. Neurol Neurochir Pol
2009;43:415–420.
e82. Kim HY, Ki CS, Kang SJ, et al. A novel LMNA gene mutation Leu162Pro and the associated clinical characteristics in a family with autosomal-dominant emery-dreifuss muscular dystrophy. Muscle Nerve 2008;38:1336–1339.
e83. Mercuri E, Brown SC, Nihoyannopoulos P, et al. Extreme variability of skeletal and cardiac muscle involvement in patients with mutations in exon 11 of the lamin A/C gene. Muscle Nerve 2005;31:602–609.
e84. Mercuri E, Counsell S, Allsop J, et al. Selective muscle involvement on magnetic resonance imaging in autosomal dominant Emery-Dreifuss muscular dystrophy. Neuropediatrics 2002;33:10–14.
e85. Mercuri E, Clements E, Offiah A, et al. Muscle magnetic resonance imaging involvement in muscular dystrophies with rigidity of the spine. Ann Neurol 2010;67:201–208.
e86. Mercuri E, Poppe M, Quinlivan R, et al. Extreme variability of phenotype in patients with an identical missense mutation in the lamin A/C gene: from congenital onset with severe phenotype to milder classic Emery-Dreifuss variant. Arch Neurol 2004;61:690–694.
e87. Park YE, Hayashi YK, Goto K, et al. Nuclear changes in skeletal muscle extend to satellite cells in autosomal dominant Emery-Dreifuss muscular dystrophy/limb-girdle muscular dystrophy 1B. Neuromuscul Disord 2009;19:29–36.
e88. Rudenskaya GE, Polyakov AV, Tverskaya SM, et al. Laminopathies in Russian families.
Clin Genet 2008;74:127–133.
e89. Sanna T, Dello Russo A, Toniolo D, et al. Cardiac features of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutations. Eur Heart J 2003;24:2227–2236.
e90. Sewry CA, Brown SC, Mercuri E, et al. Skeletal muscle pathology in autosomal dominant Emery-Dreifuss muscular dystrophy with lamin A/C mutations. Neuropathol Appl
Neurobiol 2001;27:281–290.
e91. Smith GC, Kinali M, Prasad SK, et al. Primary myocardial dysfunction in autosomal dominant EDMD. A tissue Doppler and cardiovascular magnetic resonance study. J Cardiovasc Magn Reson 2006;8:723–730.
e92. van der Kooi AJ, Ledderhof TM, de Voogt WG, et al. A newly recognized autosomal dominant limb girdle muscular dystrophy with cardiac involvement. Ann Neurol
1996;39:636–642.
e93. Walter MC, Witt TN, Weigel BS, et al. Deletion of the LMNA initiator codon leading to a neurogenic variant of autosomal dominant Emery-Dreifuss muscular dystrophy.
Neuromuscul Disord 2005;15:40–44.
e94. Ambrosi P, Mouly-Bandini A, Attarian S, Habib G. Heart transplantation in 7 patients from a single family with limb-girdle muscular dystrophy caused by lamin A/C mutation. Int J
Cardiol 2009;137:e75–e76.
e95. Antoniades L, Eftychiou C, Kyriakides T, Christodoulou K, Katritsis DG. Malignant mutation in the lamin A/C gene causing progressive conduction system disease and early sudden death in a family with mild form of limb-girdle muscular dystrophy. J Interv Card Electrophysiol 2007;19:1–7.
e96. Bécane H-M, Bonne G, Varnous S, et al. High incidence of sudden death with conduction system and myocardial disease due to lamins A and C gene mutation. Pacing Clin Electrophysiol 2000;23:1661–1666.
e97. Brodsky GL, Muntoni F, Miocic S, Sinagra G, Sewry C, Mestroni L. Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation 2000;101:473–476.
e98. Brown CA, Lanning RW, McKinney KQ, et al. Novel and recurrent mutations in lamin A/C in patients with Emery-Dreifuss muscular dystrophy. Am J Med Genet 2001;102:359– 367.
e99. Carboni N, Mura M, Marrosu G, et al. Muscle MRI findings in patients with an apparently exclusive cardiac phenotype due to a novel LMNA gene mutation. Neuromuscul Disord 2008; 18:291–298.
e100. Chang SH, Tsai CT, Lai LP, Lei MH. Identification of a lamin A/C gene mutation in a Taiwanese family with limb girdle muscular dystrophy and cardiomyopathy. Int J Cardiol 2010;145:598–599.
e101. Charniot JC, Pascal C, Bouchier C, et al. Functional consequences of an LMNA mutation associated with a new cardiac and non-cardiac phenotype. Hum Mutat 2003;21:473–481.
e102. Felice KJ, Schwartz RC, Brown CA, Leicher CR, Grunnet ML. Autosomal dominant
Emery-Dreifuss dystrophy due to mutations in rod domain of the lamin A/C gene. Neurology 2000;55:275–280.
e103. Granger B, Gueneau L, Drouin-Garraud V, et al. Modifier locus of the skeletal muscle involvement in Emery-Dreifuss muscular dystrophy. Hum Genet 2011;129:149–159.
e104. Jakobs PM, Hanson EL, Crispell KA, et al. Novel lamin A/C mutations in two families with dilated cardiomyopathy and conduction system disease. J Card Fail 2001;7:249–256.
e105. Jimenez-Escrig A, Gobernado I, Garcia-Villanueva M, Sanchez-Herranz A. Autosomal recessive Emery-Dreifuss muscular dystrophy caused by a novel mutation (R225Q) in the lamin A/C gene identified by exome sequencing. Muscle Nerve 2012;45:605–610.
e106. Komaki H, Hayashi YK, Tsuburaya R, et al. Inflammatory changes in infantile-onset
LMNA-associated myopathy. Neuromuscul Disord 2011;21:563–568.
e107. Maioli MA, Marrosu G, Mateddu A, et al. A novel mutation in the central rod domain of lamin A/C producing a phenotype resembling the Emery-Dreifuss muscular dystrophy phenotype. Muscle Nerve 2007;36:828–832.
e108. Menezes M, Waddell LB, Evesson FJ, et al. Importance and challenge of making an early diagnosis in LMNA-related muscular dystrophy. Neurology 2012;78:1258–1263.
e109. Meune C, Khouzami L, Wahbi K, et al. Blood glutathione decrease in subjects carrying lamin A/C gene mutations is an early marker of cardiac involvement. Neuromuscul Disord 2012;22:252–257.
e110. Mittelbronn M, Hanisch F, Gleichmann M, et al. Myofiber degeneration in autosomal dominant Emery-Dreifuss muscular dystrophy (AD-EDMD) (LGMD1B). Brain Pathol 2006;16:266–272.
e111. Pasotti M, Klersy C, Pilotto A, et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol 2008;52:1250–1260.
e112. Rankin J, Auer-Grumbach M, Bagg W, et al. Extreme phenotypic diversity and nonpenetrance in families with the LMNA gene mutation R644C. Am J Med Genet A 2008;146A:1530–1542.
e113. Sabatelli P, Lattanzi G, Ognibene A, et al. Nuclear alterations in autosomal-dominant
Emery-Dreifuss muscular dystrophy. Muscle Nerve 2001;24:826–829.
e114. Spuler S, Kalbhenn T, Zabojszcza J, et al. Muscle and nerve pathology in Dunnigan familial partial lipodystrophy. Neurology 2007;68:677–683.
e115. van der Kooi AJ, Bonne G, Eymard B, et al. Lamin A/C mutations with lipodystrophy, cardiac abnormalities, and muscular dystrophy. Neurology 2002;59:620–623.
e116. van der Kooi AJ, van Meegen M, Ledderhof TM, McNally EM, de Visser M, Bolhuis PA. Genetic localization of a newly recognized autosomal dominant limb-girdle muscular dystrophy with cardiac involvement (LGMD1B) to chromosome 1q11-21. Am J Hum Genet 1997;60:891–895.
e117. Vantyghem M, Pigny P, Maurage CA, et al. Patients with familial partial lipodystrophy of the Dunnigan type due to a LMNA R482W mutation show muscular and cardiac abnormalities. J Clin Endocrinol Metab 2004;89:5337–5346.
e118. Yuan WL, Huang CY, Wang JF, et al. R25G mutation in exon 1 of LMNA gene is associated with dilated cardiomyopathy and limb-girdle muscular dystrophy 1B. Chin Med J (Engl) 2009;122:2840–2845.
e119. De Sandre-Giovannoli A, Chaouch M, Kozlov S, et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet 2002;70:726–736.
e120. Minetti C, Sotgia F, Bruno C, et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet 1998;18(4):365–368.
e121. Aboumousa A, Hoogendijk J, Charlton R, et al. Caveolinopathy--new mutations and additional symptoms. Neuromuscul Disord 2008;18(7):572–578.
e122. González-Pérez P, Gallano P, González-Quereda L, et al. Phenotypic variability in a
Spanish family with a Caveolin-3 mutation. J Neurol Sci 2009;276:95–98.
e123. Cagliani R, Bresolin N, Prelle A, et al. A CAV3 microdeletion differentially affects skeletal muscle and myocardium. Neurology 2003;61:1513–1519.
e124. Catteruccia, M, Sanna T, Santorelli FM, et al. Rippling muscle disease and cardiomyopathy associated with a mutation in the CAV3 gene. Neuromuscul Disord 2009;19:779–783.
e125. Dotti MT, Malandrini A, Gambelli S, Salvadori C, De Stefano N, Federico A. A new missense mutation in caveolin-3 gene causes rippling muscle disease. J Neurol Sci
2006;243:61–64.
e126. Fischer D, Schroers A, Blümcke I, et al. Consequences of a novel caveolin-3 mutation in a large German family. Ann Neurol 2003;53:233–241.
e127. Fulizio L, Nascimbeni AC, Fanin M, et al. Molecular and muscle pathology in a series of caveolinopathy patients. Hum Mutat 2005;25:82–89.
e128. Jacobi C, Ruscheweyh R, Vorgerd M, Weber MA, Storch-Hagenlocher B, Meinck HM.
Rippling muscle disease: variable phenotype in a family with five afflicted members.
Muscle Nerve 2010;41:128–132.
e129. Ricker K, Moxley RT, Rohkamm R. Rippling muscle disease. Arch Neurol 1989;46:405–
408.
e130. Sundblom J, Stålberg E, Osterdahl M, et al. Bedside diagnosis of rippling muscle disease in CAV3 p.A46T mutation carriers. Muscle Nerve 2010;41:751–757.
e131. Vorgerd M, Bolz H, Patzold T, Kubisch C, Malin JP, Mortier W. Phenotypic variability in rippling muscle disease. Neurology 1999;52:1453–1459.
e132. Yabe I, Kawashima A, Kikuchi S, et al. Caveolin-3 gene mutation in Japanese with rippling muscle disease. Acta Neurol Scand 2003;108:47–51.
e133. Hackman P, Sandell S, Sarparanta J, et al. Four new Finnish families with LGMD1D; refinement of the clinical phenotype and the linked 7q36 locus. Neuromuscul Disord 2011;21:338–344.
e134. Sarparanta J, Jonson PH, Golzio C, et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat Genet 2012;44:450–455, S1–S2.
e135. Harms MB, Sommerville RB, Allred P, et al. Exome sequencing reveals DNAJB6 mutations in dominantly-inherited myopathy. Ann Neurol 2012;71:407–416.
e136. Urtasun M, Sáenz A, Roudaut C, et al. Limb-girdle muscular dystrophy in Guipúzcoa
(Basque Country, Spain). Brain 1998;121:1735–1747.
e137. Angelini C, Nardetto L, Borsato C, et al. The clinical course of calpainopathy (LGMD2A) and dysferlinopathy (LGMD2B). Neurol Res 2010;32:41–46.
e138. Balci B, Aurino S, Haliloglu G, et al. Calpain-3 mutations in Turkey. Eur J Pediatr 2006;
165:293–298.
e139. Beckmann JS, Richard I, Hillaire D, et al. A gene for limb-girdle muscular dystrophy maps to chromosome 15 by linkage. C R Acad Sci III 1991;312:141–148.
e140. Blázquez L, Azpitarte M, Sáenz A, et al. Characterization of novel CAPN3 isoforms in white blood cells: an alternative approach for limb-girdle muscular dystrophy 2A diagnosis. Neurogenetics 2008;9:173–182.
e141. Charlton R, Henderson M, Richards J, et al. Immunohistochemical analysis of calpain 3:
advantages and limitations in diagnosing LGMD2A. Neuromuscul Disord 2009;19:449– 457.
e142. Chrobáková T, Hermanová M, Kroupová I, et al. Mutations in Czech LGMD2A patients revealed by analysis of calpain3 mRNA and their phenotypic outcome. Neuromuscul Disord 2004;14:659–665.
e143. de Paula F, Vainzof M, Passos-Bueno MR, et al. Clinical variability in calpainopathy: what makes the difference? Eur J Hum Genet 2002;10:825–832.
e144. Degardin A, Morillon D, Lacour A, Cotten A, Vermersch P, Stojkovic T. Morphologic imaging in muscular dystrophies and inflammatory myopathies. Skeletal Radiol 2010;39(12):1219–1227.
e145. Fanin M, Nardetto L, Nascimbeni AC, et al. Correlations between clinical severity, genotype and muscle pathology in limb girdle muscular dystrophy type 2A. J Med Genet 2007;44(10):609–614.
e146. Fanin M, Nascimbeni AC, Angelini C. Screening of calpain-3 autolytic activity in LGMD muscle: a functional map of CAPN3 gene mutations. J Med Genet 2007;44:38–43.
e147. Fanin M, Pegoraro E, Matsuda-Asada C, Brown RH Jr, Angelini C. Calpain-3 and dysferlin protein screening in patients with limb-girdle dystrophy and myopathy. Neurology 2001;56:660–665.
e148. Fardeau M, Hillaire D, Mignard C, et al. Juvenile limb-girdle muscular dystrophy. Clinical, histopathological and genetic data from a small community living in the Reunion Island. Brain 1996;119:295–308.
e149. Fischer D, Walter MC, Kesper K, et al. Diagnostic value of muscle MRI in differentiating
LGMD2I from other LGMDs. J Neurol 2005;252:538–547.
e150. Groen E, Charlton R, Barresi R, et al. Analysis of the UK diagnostic strategy for limb girdle muscular dystrophy 2A. Brain 2007;130:3237–3249.
e151. Kawai H, Akaike M, Kunishige M, et al. Clinical, pathological, and genetic features of limb-girdle muscular dystrophy type 2A with new calpain 3 gene mutations in seven patients from three Japanese families. Muscle Nerve 1998;21:1493–1501.
e152. Mercuri E, Bushby K, Ricci E, et al. Muscle MRI findings in patients with limb girdle muscular dystrophy with calpain 3 deficiency (LGMD2A) and early contractures. Neuromuscul Disord 2005;15:164–171.
e153. Minami N, Nishino I, Kobayashi O, Ikezoe K, Goto Y, Nonoka I. Mutations of calpain 3 gene in patients with sporadic limb-girdle muscular dystrophy in Japan. J Neurol Sci 1999;171:31–37.
e154. Schessl J, Walter MC, Schreiber G, et al. Phenotypic variability in siblings with calpainopathy (LGMD2A). Acta Myol 2008;27:54–58.
e155. Stramare R, Beltrame V, Dal Borgo R, et al. MRI in the assessment of muscular pathology: a comparison between limb-girdle muscular dystrophies, hyaline body myopathies and myotonic dystrophies. Radiol Med 2010;115:585–599.
e156. Sveen ML, Thune JJ, Køber L, Vissing J. Cardiac involvement in patients with limb-girdle muscular dystrophy type 2 and Becker muscular dystrophy. Arch Neurol 2008;65:1196–1201.
e157. Topaloğlu H, Dinçer P, Richard I, et al. Calpain-3 deficiency causes a mild muscular dystrophy in childhood. Neuropediatrics 1997;28:212–216.
e158. Pollitt C, Anderson LV, Pogue R, Davison K, Pyle A, Bushby KM. The phenotype of calpainopathy: diagnosis based on a multidisciplinary approach. Neuromuscul Disord
2001;11:287–296.
e159. Starling A, de Paula F, Silva H, Vainzof M, Zatz M. Calpainopathy: how broad is the spectrum of clinical variability? J Mol Neurosci 2003;21:233–236.
e160. Amato AA. Adults with eosinophilic myositis and calpain-3 mutations. Neurology
2008;70:730–731.
e161. Choi ER, Park SJ, Choe YH, et al. Early detection of cardiac involvement in Miyoshi myopathy: 2D strain echocardiography and late gadolinium enhancement cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2010;12:31.
e162. Aoki M, Liu J, Richard I, et al. Genomic organization of the dysferlin gene and novel mutations in Miyoshi myopathy. Neurology 2001;57:271–278.
e163. Argov Z, Sadeh M, Mazor K, et al. Muscular dystrophy due to dysferlin deficiency in
Libyan Jews. Clinical and genetic features. Brain 2000;123:1229–1237.
e164. Comerlato EA, Scola RH, Werneck LC. Limb-girdle muscular dystrophy: an immunohistochemical diagnostic approach. Arq Neuropsiquiatr 2005;63(2A):235–245.
e165. Cupler EJ, Bohlega S, Hessler R, McLean D, Stigsby B, Ahmad J. Miyoshi myopathy in Saudi Arabia: clinical, electrophysiological, histopathological and radiological features. Neuromuscul Disord 1998;8:321–326.
e166. Fanin M, Angelini C. Muscle pathology in dysferlin deficiency. Neuropathol Appl
Neurobiol 2002;28:461–470.
e167. Khadilkar S, Singh RK, Kulkarni KS, Chitale AR. A study of clinical and laboratory features of 14 Indian patients with dysferlinopathy. J Clin Neuromuscul Dis 2004;6:1–8.
e168. Klinge L, Aboumousa A, Eagle M, et al. New aspects on patients affected by dysferlin deficient muscular dystrophy. J Neurol Neurosurg Psychiatry 2010;81:946–953.
e169. McNally EM, Ly CT, Rosenmann H, et al. Splicing mutation in dysferlin produces limbgirdle muscular dystrophy with inflammation. Am J Med Genet 2000;91:305–312.
e170. Pradhan S. Clinical and magnetic resonance imaging features of ‘diamond on quadriceps’ sign in dysferlinopathy. Neurol India 2009;57:172–175.
e171. Prelle A, Sciacco M, Tancredi L, et al. Clinical, morphological and immunological evaluation of six patients with dysferlin deficiency. Acta Neuropathol 2003;105:537–542.
e172. Ro LS, Lee-Chen GJ, Lin TC, et al. Phenotypic features and genetic findings in 2 chinese families with Miyoshi distal myopathy. Arch Neurol 2004;61:1594–1599.
e173. Rosales XQ, Gastier-Foster JM, Lewis S, et al. Novel diagnostic features of dysferlinopathies. Muscle Nerve 2010;42:14–21.
e174. Takahashi T, Aoki M, Tateyama M, et al. Dysferlin mutations in Japanese Miyoshi myopathy: relationship to phenotype. Neurology 2003;60:1799–1804.
e175. Ueyama H, Kumamoto T, Nagao S, et al. A new dysferlin gene mutation in two Japanese families with limb-girdle muscular dystrophy 2B and Miyoshi myopathy. Neuromuscul Disord 2001;11:139–145.
e176. Vainzof M, Anderson LV, McNally EM, et al. Dysferlin protein analysis in limb-girdle muscular dystrophies. J Mol Neurosci 2001;17:71–80.
e177. Vilchez JJ, Gallano P, Gallardo E, et al. Identification of a novel founder mutation in the DYSF gene causing clinical variability in the Spanish population. Arch Neurol 2005;62:1256–1259.
e178. Weiler T, Greenberg CR, Nylen E, et al. Limb-girdle muscular dystrophy and Miyoshi myopathy in an aboriginal Canadian kindred map to LGMD2B and segregate with the same haplotype. Am J Hum Genet 1996;59:872–878.
e179. Anderson LV, Harrison RM, Pogue R, et al. Secondary reduction in calpain 3 expression in patients with limb girdle muscular dystrophy type 2B and Miyoshi myopathy (primary dysferlinopathies). Neuromuscul Disord 2000;10:553–559.
e180. Angelini C, Peterle E, Gaiani A, Bortulussi L, Borsato C. Dysferlinopathy course and sportive activity: clues for possible treatment. Acta Myol 2011;30:127–132.
e181. Brunn A, Schröder R, Deckert M. The inflammatory reaction pattern distinguishes primary dysferlinopathies from idiopathic inflammatory myopathies: an important role for the membrane attack complex. Acta Neuropathol 2006;112:325–332.
e182. Cagliani R, Fortunato F, Giorda R, et al. Molecular analysis of LGMD-2B and MM patients: identification of novel DYSF mutations and possible founder effect in the Italian population. Neuromuscul Disord 2003;13:788–795.
e183. Cagliani R, Magri F, Toscano A, et al. Mutation finding in patients with dysferlin deficiency and role of the dysferlin interacting proteins annexin A1 and A2 in muscular dystrophies. Hum Mutat 2005;26:283.
e184. Cenacchi G, Fanin M, De Giorgi LB, Angelini C. Ultrastructural changes in dysferlinopathy support defective membrane repair mechanism. J Clin Pathol 2005;58:190–195.
e185. Leshinsky-Silver E, Argov Z, Rozenboim L, et al. Dysferlinopathy in the Jews of the Caucasus: a frequent mutation in the dysferlin gene. Neuromuscul Disord 2007;17:950– 954.
e186. Cho H, Sung DH, Kim EJ, Yoon CH, Ki CS, Kim JW. Clinical and genetic analysis of Korean patients with Miyoshi myopathy: identification of three novel mutations in the DYSF gene. J Korean Med Sci 2006;21:724–727.
e187. Gallardo E, Rojas-García R, de Luna N, Pou A, Brown RH Jr, Illa I. Inflammation in dysferlin myopathy: immunohistochemical characterization of 13 patients. Neurology 2001;57:2136–2138.
e188. Gayathri N, Alefia R, Nalini A, et al. Dysferlinopathy: spectrum of pathological changes in skeletal muscle tissue. Indian J Pathol Microbiol 2011;54:350–354.
e189. Paradas C, Llauger J, Diaz-Manera J, et al. Redefining dysferlinopathy phenotypes based on clinical findings and muscle imaging studies. Neurology 2010;75:316–323.
e190. Kawabe K, Goto K, Nishino I, Angelini C, Hayashi YK. Dysferlin mutation analysis in a group of Italian patients with limb-girdle muscular dystrophy and Miyoshi myopathy. Eur J Neurol 2004;11:657–661.
e191. Kesper K, Kornblum C, Reimann J, Lutterbey G, Schröder R, Wattjes MP. Pattern of skeletal muscle involvement in primary dysferlinopathies: a whole-body 3.0-T magnetic resonance imaging study. Acta Neurol Scand 2009;120:111–118.
e192. Nguyen K, Bassez G, Bernard R, et al. Dysferlin mutations in LGMD2B, Miyoshi myopathy, and atypical dysferlinopathies. Hum Mutat 2005;26:165.
e193. Park YE, Kim HS, Lee CH, Nam TS, Choi YC, Kim DS. Two common mutations
(p.GIn832X and c.663+1G>C) account for about a third of the DYSF mutations in Korean patients with dysferlinopathy. Neuromuscul Disord 2012;22:505–510.
e194. Piccolo F, Moore SA, Ford GC, Campbell KP. Intracellular accumulation and reduced sarcolemmal expression of dysferlin in limb--girdle muscular dystrophies. Ann Neurol 2000;48:902–912.
e195. Pradhan S. Calf-head sign in Miyoshi myopathy. Arch Neurol 2006;63:1414–1417.
e196. Weiler T, Bashir R, Anderson LV, et al. Identical mutation in patients with limb girdle muscular dystrophy type 2B or Miyoshi myopathy suggests a role for modifier gene(s). Hum Mol Genet 1999;8:871–877.
e197. Rosales XQ, Moser SJ, Tran T, et al. Cardiovascular magnetic resonance of cardiomyopathy in limb girdle muscular dystrophy 2B and 2I. J Cardiovasc Magn Reson 2011;13:39.
e198. Saito A, Higuchi I, Nakagawa M, et al. Miyoshi myopathy patients with novel 5' splicing donor site mutations showed different dysferlin immunostaining at the sarcolemma. Acta Neuropathol 2002;104:615–620.
e199. Shunchang S, Fan Q, Huacheng W, et al. Dysferlin mutation in a Chinese pedigree with
Miyoshi myopathy. Clin Neurol Neurosurg 2006;108:369–373.
e200. Spuler S, Carl M, Zabojszcza J, et al. Dysferlin-deficient muscular dystrophy features amyloidosis. Ann Neurol 2008;63:323–328.
e201. Vernengo L, Oliveira J, Krahn M, et al. Novel ancestral Dysferlin splicing mutation which migrated from the Iberian peninsula to South America. Neuromuscul Disord 2011;21:328–337.
e202. Liewluck T, Pongpakdee S, Witoonpanich R, et al. Novel DYSF mutations in Thai patients with distal myopathy. Clin Neurol Neurosurg 2009;111:613–618.
e203. Brummer D, Walter MC, Palmbach M, et al. Long-term MRI and clinical follow-up of symptomatic and presymptomatic carriers of dysferlin gene mutations. Acta Myol 2005;24:6–16.
e204. Wenzel K, Geier C, Qadri F, et al. Dysfunction of dysferlin-deficient hearts. J Mol Med (Berl) 2007;85:1203–1214.
e205. Mahjneh I, Marconi G, Bushby K, Anderson LV, Tolvanen-Mahjneh H, Somer H. Dysferlinopathy (LGMD2B): a 23-year follow-up study of 10 patients homozygous for the same frameshifting dysferlin mutations. Neuromuscul Disord 2001;11:20–26.
e206. Illarioshkin SN, Ivanova-Smolenskaya IA, Tanaka H, et al. Clinical and molecular analysis of a large family with three distinct phenotypes of progressive muscular dystrophy. Brain 1996;119:1895–1909.
e207. Illa I, Serrano-Munuera C, Gallardo E, et al. Distal anterior compartment myopathy: a dysferlin mutation causing a new muscular dystrophy phenotype. Ann Neurol 2001;49:130–134.
e208. Kefi M, Amouri R, Driss A, et al. Phenotype and sarcoglycan expression in Tunisian LGMD 2C patients sharing the same del521-T mutation. Neuromuscul Disord 2003;13:779–787.
e209. Todorova A, Tournev I, Ninova N, Georgieva V, Kremensky I. Screening for C283Y gamma-sarcoglycan mutation in a high-risk group of Bulgarian Gypsies: evidence for a geographical localization and a non-random distribution among Gypsy subgroups. Community Genet 2002;5:217–221.
e210. Bönnemann CG, Wong J, Jones KJ, et al. Primary gamma-sarcoglycanopathy (LGMD 2C): broadening of the mutational spectrum guided by the immunohistochemical profile. Neuromuscul Disord 2002;12:273–280.
e211. Calvo F, Teijeira S, Fernandez JM, et al. Evaluation of heart involvement in gammasarcoglycanopathy (LGMD2C). A study of ten patients. Neuromuscul Disord 2000;10:560–566.
e212. Duncan D, Kang PB, Rabbat JC, et al. A novel mutation in two families with limb-girdle muscular dystrophy type 2C. Neurology 2006;67:167–169.
e213. Klinge L, Dekomien G, Aboumousa A, et al. Sarcoglycanopathies: can muscle immunoanalysis predict the genotype? Neuromuscul Disord 2008;18(12):934–941.
e214. Lasa A, Piccolo F, de Diego C, et al. Severe limb girdle muscular dystrophy in Spanish gypsies: further evidence for a founder mutation in the gamma-sarcoglycan gene. Eur J Hum Genet 1998;6:396–399.
e215. Leal GF, da-Silva EO. Limb-girdle muscular dystrophy with apparently different clinical courses within sexes in a large inbred kindred. J Med Genet 1999;36:714–718.
e216. Merlini L, Kaplan JC, Navarro C, et al. The limb-girdle muscular dystrophy 2C in Gypsies. Acta Myol 2001;20:188–191.
e217. van der Kooi AJ, de Visser M, van Meegen M, et al. A novel gamma-sarcoglycan mutation causing childhood onset, slowly progressive limb girdle muscular dystrophy. Neuromuscul Disord 1998;8:305–308.
e218. Melacini P, Fanin M, Duggan DJ, et al. Heart involvement in muscular dystrophies due to sarcoglycan gene mutations. Muscle Nerve 1999;22:473–479.
e219. Vainzof M, Passos-Bueno MR, Canovas M, et al. The sarcoglycan complex in the six autosomal recessive limb-girdle muscular dystrophies. Hum Mol Genet 1996;5:1963– 1969.
e220. Spengos K, Walter MC, Dekomien G, Papadopoulos K, Lochmüller H, Manta P. C283Y mutation in the gamma-sarcoglycan gene in Greek Gypsies with severe limb girdle muscular dystrophy. Eur J Neurol 2010;17:e41–e42.
e221. Eymard B, Romero NB, Leturcq F, et al. Primary adhalinopathy (alphasarcoglycanopathy): clinical, pathologic, and genetic correlation in 20 patients with autosomal recessive muscular dystrophy. Neurology 1997;48(5):1227–1234.
e222. Vlak M, van der Kooi E, Angelini C. Correlation of clinical function and muscle CT scan images in limb-girdle muscular dystrophy. Neurol Sci 2000;21(5 Suppl):S975–S977.
e223. Hackman P, Juvonen V, Saraparanta J, et al. Enrichment of the R77C alpha-sarcoglycan gene mutation in Finnish LGMD2D patients. Muscle Nerve 2005;31(2):199–204.
e224. Fendri K, Kefi M, Hentati F, Amouri R. Genetic heterogeneity within a consanguineous family involving the LGMD 2D and the LGMD 2C genes. Neuromuscul Disord 2006;16(5):316–320.
e225. Vainzof M, Moreira ES, Canovas M, et al. Partial alpha-sarcoglycan deficiency with retention of the dystrophin-glycoprotein complex in a LGMD2D family. Muscle Nerve 2000;23:984–988.
e226. Piccolo F, Roberds SL, Jeanpierre M, et al. Primary adhalinopathy: a common cause of autosomal recessive muscular dystrophy of variable severity. Nat Genet 1995;10:243– 245.
e227. Tétreault M, Srour M, Allyson J, et al. Founder mutation for α-sarcoglycan-LGMD2D in a
Magdalen Islands Acadian cluster. Can J Neurol Sci 2011;38:747–752.
e228. Balci B, Wilichowski E, Haliloğlu G, et al. Beta-sarcoglycan gene mutations in Turkey.
Acta Myol 2004;23(3):154–158.
e229. Bönnemann CG, Wong J, Ben Hamida C, Hamida MB, Hentati F, Kunkel LM. LGMD 2E in Tunisia is caused by a homozygous missense mutation in beta-sarcoglycan exon 3.
Neuromuscul Disord 1998;8:193–197.
e230. Fanin M, Melacini P, Boito C, Pegoraro E, Angelini C. LGMD2E patients risk developing dilated cardiomyopathy. Neuromuscul Disord 2003;13:303–309.
e231. Nigro V, de Sá Moreira E, Piluso G, et al. Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the delta-sarcoglycan gene. Nat Genet 1996;14:195–198.
e232. Moreira ES, Vainzof M, Marie SK, Sertié AL, Zatz M, Passos-Bueno MR. The seventh form of autosomal recessive limb-girdle muscular dystrophy is mapped to 17q11-12. Am J Hum Genet 1997;61:151–159.
e233. Moreira ES, Wiltshire TJ, Faulkner G, et al. Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nat Genet 2000;24(2):163–166.
e234. Schoser BG, Frosk P, Engel AG, Klutzny U, Lochmüller H, Wrogemann K. Commonality of TRIM32 mutation in causing sarcotubular myopathy and LGMD2H. Ann Neurol 2005;57(4):591–595.
e235. Borg K, Stucka R, Locke M, et al. Intragenic deletion of TRIM32 in compound heterozygotes with sarcotubular myopathy/LGMD2H. Hum Mutat 2009;30(9):E831–E844.
e236. Saccone V, Palmieri M, Passamano L, et al. Mutations that impair interaction properties of TRIM32 associated with limb-girdle muscular dystrophy 2H. Hum Mutat 2008;29:240– 247.
e237. Weiler T, Greenberg CR, Zelinski T, et al. A gene for autosomal recessive limb-girdle muscular dystrophy in Manitoba Hutterites maps to chromosome region 9q31-q33: evidence for another limb-girdle muscular dystrophy locus. Am J Hum Genet 1998;63:140–147.
e238. dePaula F, Vieira N, Starling A, et al. Asymptomatic carriers for homozygous novel mutations in the FKRP gene: the other end of the spectrum. Eur J Hum Genet 2003;11:923–930.
e239. Gaul C, Deschauer M, Tempelmann C, et al. Cardiac involvement in limb-girdle muscular dystrophy 2I: conventional cardiac diagnostic and cardiovascular magnetic resonance. J Neurol 2006;253:1317–1322.
e240. Bourteel H, Vermersch P, Cuisset JM, et al. Clinical and mutational spectrum of limbgirdle muscular dystrophy type 2I in 11 French patients. J Neurol Neurosurg Psychiatry 2009;80:1405–1408.
e241. Brown SC, Torelli S, Brockington M, et al. Abnormalities in alpha-dystroglycan expression in MDC1C and LGMD2I muscular dystrophies. Am J Pathol 2004;164:727– 737.
e242. Driss A, Amouri R, Ben Hamida C, et al. A new locus for autosomal recessive limb-girdle muscular dystrophy in a large consanguineous Tunisian family maps to chromosome 19q13.3. Neuromuscul Disord 2000;10:240–246.
e243. Harel T, Goldberg Y, Shalev SA, Chervinski I, Ofir R, Birk OS. Limb-girdle muscular dystrophy 2I: phenotypic variability within a large consanguineous Bedouin family associated with a novel FKRP mutation. Eur J Hum Genet 2004;12:38–43.
e244. Jimenez-Mallebrera C, Torelli S, Feng L, et al. A comparative study of alpha-dystroglycan glycosylation in dystroglycanopathies suggests that the hypoglycosylation of alpha-dystroglycan does not consistently correlate with clinical severity. Brain Pathol 2009;19:596–611.
e245. Poppe M, Cree L, Bourke J, et al. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology 2003;60:1246–1251.
e246. Sveen ML, Jeppesen TD, Hauerslev S, Krag TO, Vissing J. Endurance training: an effective and safe treatment for patients with LGMD2I. Neurology 2007;68(1):59–61.
e247. Wahbi K, Meune C, Hamouda el H, et al. Cardiac assessment of limb-girdle muscular dystrophy 2I patients: an echography, Holter ECG and magnetic resonance imaging study. Neuromuscul Disord 2008;18:650–655.
e248. Kefi M, Amouri R, Chabrak S, Mechmeche R, Hentati F. Variable cardiac involvement in
Tunisian siblings harboring FKRP gene mutations. Neuropediatrics 2008;39:113–115.
e249. Frosk P, Greenberg CR, Tennese AA, et al. The most common mutation in FKRP causing limb girdle muscular dystrophy type 2I (LGMD2I) may have occurred only once and is present in Hutterites and other populations. Hum Mutat 2005;25:38–44.
e250. Lindberg C, Sixt C, Oldfors A. Episodes of exercise-induced dark urine and myalgia in
LGMD 2I. Acta Neurol Scand 2012;125:285–287.
e251. Mathews KD, Stephan CM, Laubenthal K, et al. Myoglobinuria and muscle pain are common in patients with limb-girdle muscular dystrophy 2I. Neurology 2011;76:194–195.
e252. Müller T, Krasnianski M, Witthaut R, Deschauer M, Zierz S. Dilated cardiomyopathy may be an early sign of the C826A Fukutin-related protein mutation. Neuromuscul Disord 2005;15:372–376.
e253. Palmieri A, Manara R, Bello L, et al. Cognitive profile and MRI findings in limb-girdle muscular dystrophy 2I. J Neurol 2011;258:1312–1320.
e254. Vieira NM, Schlesinger D, de Paula F, Vainzof M, Zatz M. Mutation analysis in the FKRP gene provides an explanation for a rare cause of intrafamilial clinical variability in LGMD2I. Neuromuscul Disord 2006;16:870–873.
e255. Yilmaz A, Gdynia HJ, Ponfick M, Ludolph AC, Rösch S, Sechtem U. The proteoglycandystrophin complex in genetic cardiomyopathies--lessons from three siblings with limbgirdle muscular dystrophy-2I (LGMD-2I). Clin Res Cardiol 2011;100:611–615.
e256. Mercuri E, Brockington M, Straub V, et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann Neurol 2003;53:537–542.
e257. Walter MC, Petersen JA, Stucka R, et al. FKRP (826C>A) frequently causes limb-girdle muscular dystrophy in German patients. J Med Genet 2004;41:e50.
e258. Udd B. Limb-girdle type muscular dystrophy in a large family with distal myopathy: homozygous manifestation of a dominant gene? J Med Genet 1992;29(6):383–389.
e259. Udd B, Rapola J, Nokelainen P, Arikawa E, Somer H. Nonvacuolar myopathy in a large family with both late adult onset distal myopathy and severe proximal muscular dystrophy. J Neurol Sci 1992;113(2):214–221.
e260. Udd B, Partanen J, Halonen P, et al. Tibial muscular dystrophy. Late adult-onset distal myopathy in 66 Finnish patients. Arch Neurol 1993;50(6):604-608.
e261. Udd B, Haravuori H, Kalimo H, et al. Tibial muscular dystrophy--from clinical description to linkage on chromosome 2q31. Neuromuscul Disord 1998;8(5):327–332.
e262. Van den Bergh PY, Bouquiaux O, Verellen C, et al. Tibial muscular dystrophy in a Belgian family. Ann Neurol 2003;54(2):248–251.
e263. Mahjneh I, Lamminen AE, Udd B, et al. Muscle magnetic resonance imaging shows distinct diagnostic patterns in Welander and tibial muscular dystrophy. Acta Neurol Scand 2004;110(2):87–93.
e264. Pénisson-Besnier I, Hackman P, Suominen T, et al. Myopathies caused by homozygous titin mutations: limb-girdle muscular dystrophy 2J and variations of phenotype. J Neurol Neurosurg Psychiatry 2010;81(11):1200–1202.
e265. Ohlsson M, Hedberg C, Brådvik B, et al. Hereditary myopathy with early respiratory failure associated with a mutation in A-band titin. Brain 2012;135(Pt 6):1682–1694.
e266. Pfeffer G, Elliott HR, Griffin H, et al. Titin mutation segregates with hereditary myopathy with early respiratory failure. Brain 2012;135(Pt 6):1695–1713.
e267. Carmignac V, Salih MA, Quijano-Roy S, et al. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol 2007;61:340–351.
e268. Udd B, Kääriänen H, Somer H. Muscular dystrophy with separate clinical phenotypes in a large family. Muscle Nerve 1991;14:1050–1058.
e269. Bolduc V, Marlow G, Boycott KM, et al. Recessive mutations in the putative calciumactivated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am J Hum Genet 2010;86(2):213–221.
e270. Mahjneh I, Jaiswal J, Lamminen A, et al. A new distal myopathy with mutation in anoctamin 5. Neuromuscul Disord 2010;20(12):791–795.
e271. Schessl J, Kress W, Schoser B. Novel ANO5 mutations causing hyper-CK-emia, limb girdle muscular weakness and Miyoshi type of muscular dystrophy. Muscle Nerve 2012;45:740–742.
e272. Jaiswal JK, Marlow G, Summerill G, et al. Patients with a non-dysferlin Miyoshi myopathy have a novel membrane repair defect. Traffic 2007;8(1):77–88.
e273. Jarry J, Rioux MF, Bolduc V, et al. A novel autosomal recessive limb-girdle muscular dystrophy with quadriceps atrophy maps to 11p13-p12. Brain 2007;130(Pt 2):368–380.
e274. Godfrey C, Escolar D, Brockington M, et al. Fukutin gene mutations in steroid-responsive limb girdle muscular dystrophy. Ann Neurol 2006;60(5):603–610.
e275. Biancheri R, Falace A, Tessa A, et al. POMT2 gene mutation in limb-girdle muscular dystrophy with inflammatory changes. Biochem Biophys Res Commun 2007;363(4):1033–1037.
e276. Clement EM, Godfrey C, Tan J, et al. Mild POMGnT1 mutations underlie a novel limbgirdle muscular dystrophy variant. Arch Neurol 2008;65(1):137–141.
e277. Hara Y, Balci-Hayta B, Yoshida-Moriguchi T, et al. A dystroglycan mutation associated with limb-girdle muscular dystrophy. N Eng J Med 2011;364:939–946.
e278. Dinçer P, Balci B, Yuva Y, et al. A novel form of recessive limb girdle muscular dystrophy with mental retardation and abnormal expression of α-dystroglycan. Neuromuscul Disord 2003;13:771–778.
e279. Banwell BL, Russel J, Fukudome T, Shen XM, Stilling G, Engel AG. Myopathy, myasthenic syndrome, and epidermolysis bullosa simplex due to plectin deficiency. J Neuropathol Exp Neurol 1999;58:832–846.
e280. Charlesworth A, Chiaverini C, Chevrant-Breton J, et al. Epidermolysis bullosa simplex with PLEC mutations: new phenotypes and new mutations [published online ahead of print Jan 5, 2013]. Br J Dermatol doi: 10.1111/bjd.12202.
e281. Chavanas S, Pulkkinen L, Gache Y, et al. A homozygous nonsense mutation in the PLEC1 gene in patients with epidermolysis bullosa simplex with muscular dystrophy. J Clin Invest 1996;98:2196–2200.
e282. Forrest K, Mellerio JE, Robb S, et al. Congenital muscular dystrophy, myasthenic symptoms and epidermolysis bullosa simplex (EBS) associated with mutations in the PLEC1 gene encoding plectin. Neuromuscul Disord 2010;20:709–711.
e283. Gache Y, Chavanas S, Lacour JP, et al. Defective expression of plectin/HD1 in epidermolysis bullosa simplex with muscular dystrophy. J Clin Invest 1996;97:2289– 2298.
e284. Mellerio JE, Smith FJ, McMillian JR, et al. Recessive epidermolysis bullosa simplex associated with plectin mutations: infantile respiratory complications in two unrelated cases. Br J Dermatol 1997;137:898–906.
e285. Natsuga K, Nishie W, Shinkuma S, et al. Plectin deficiency leads to both muscular dystrophy and pyloric atresia in epidermolysis bullosa simplex. Hum Mutat 2010;31:E1687–E1698.
e286. Pulkkinen L, Smith FJ, Shimizu H, et al. Homozygous deletion mutations in the plectin gene (PLEC1) in patients with epidermolysis bullosa simplex associated with late-onset muscular dystrophy. Hum Mol Genet 1996;5:1539–1546.
e287. Rouan F, Pulkkinen L, Meneguzzi G, et al. Epidermolysis bullosa: novel and de novo premature termination codon and deletion mutations in the plectin gene predict late-onset muscular dystrophy. J Invest Dermatol 2000;114:381–387.
e288. Schara U, Tücke J, Mortier W, et al. Severe mucous membrane involvement in epidermolysis bullosa simplex with muscular dystrophy due to a novel plectin gene mutation. Eur J Pediatr 2004;163:218–222.
e289. Schröder R, Kunz WS, Rouan F, et al. Disorganization of the desmin cytoskeleton and mitochondrial dysfunction in plectin-related epidermolysis bullosa simplex with muscular dystrophy. J Neuropathol Exp Neurol 2002;61:520–530.
e290. Smith FJ, Eady RA, Leigh IM, et al. Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nat Genet 1996;13:450–457.
e291. Takizawa Y, Shimizu H, Rouan F, et al. Four novel plectin gene mutations in Japanese patients with epidermolysis bullosa with muscular dystrophy disclosed by heteroduplex scanning and protein truncation tests. J Invest Dermatol 1999;112:109–112.
e292. Yiu EM, Klausegger A, Waddell LB, et al. Epidermolysis bullosa with late-onset muscular dystrophy and plectin deficiency. Muscle Nerve 2011;44:135–141.
e293. Selcen D, Juel VC, Hobson-Webb LD, et al. Myasthenic syndrome caused by plectinopathy. Neurology 2011;76:327–336.
e294. Shimizu H, Takizawa Y, Pulkkinen L, et al. Epidermolysis bullosa simplex associated with muscular dystrophy: phenotype-genotype correlations and review of the literature. J Am Acad Dermatol 1999;41:950–956.
e295. Takahashi Y, Rouan F, Uitto J, et al. Plectin deficient epidermolysis bullosa simplex with
27-year-history of muscular dystrophy. J Dermatol Sci 2005;37:87–93.
e296. Patrizi A, Di Lernia V, Neri I, Badiali De Giorgi L, Masi M. Epidermolysis bullosa simplex associated with muscular dystrophy: a new case. Pediatr Dermatol 1994;11:342–345.
e297. Niemi KM, Sommer H, Kero M, Kanerva L, Haltia M. Epidermolysis bullosa simplex associated with muscular dystrophy with recessive inheritance. Arch Dermatol 1988;124:551–554.
e298. Natsuga K, Nishie W, Akiyama M, et al. Plectin expression patterns determine two distinct subtypes of epidermolysis bullosa simplex. Hum Mutat 2010;31:308–316.
e299. Koss-Harnes, D, Jahnsen FL, Wiche G, Soyland E, Brandtzaeg P, Gedde-Dahl T Jr. Plectin abnormality in epidermolysis bullosa simplex Ogna: non-responsiveness of basal keratinocytes to some anti-rat plectin antibodies. Exp Dermatol 1997;6:41–48.
e300. Kunz M, Rouan F, Pulkkinen L, et al. Mutation reports: epidermolysis bullosa simplex associated with severe mucous membrane involvement and novel mutations in the plectin gene. J Invest Dermatol 2000;114:376–380.
e301. Kurose K, Mori O, Hachisuka H, Shimizu H, Owaribe K, Hashimoto T. Cultured keratinocytes from plectin/HD1-deficient epidermolysis bullosa simplex showed altered ability of adhesion to the matrix. J Dermatol Sci 2000;24:184–189.
e302. McLean WH, Pulkkinen L, Smith FJ, et al. Loss of plectin causes epidermolysis bullosa with muscular dystrophy: cDNA cloning and genomic organization. Genes Dev 1996;10:1724–1735.
e303. Maselli RA, Arredondo J, Cagney O, et al. Congenital myasthenic syndrome associated with epidermolysis bullosa caused by homozygous mutations in PLEC1 and CHRNE. Clin Genet 2011;80:444–451.
e304. McMillan JR, Akiyama M, Rouan F, et al. Plectin defects in epidermolysis bullosa simplex with muscular dystrophy. Muscle Nerve 2007;35:24–35.
e305. Doriguzzi C, Palmucci L, Mongini T, et al. Congenital muscular dystrophy associated with familial junctional epidermolysis bullosa letalis. Eur Neurol 1993;33:454–460.
e306. Fine JD, Stenn J, Johnson L, Wright T, Bock HG, Horiguchi Y. Autosomal recessive epidermolysis bullosa simplex. Generalized phenotypic features suggestive of junctional or dystrophic epidermolysis bullosa, and association with neuromuscular diseases. Arch Dermatol 1989;125:931–938.
e307. Bolling MC, Pas HH, de Visser M, et al. PLEC1 mutations underlie adult-onset dilated cardiomyopathy in epidermolysis bullosa simplex with muscular dystrophy. J Invest Dermatol 2010;130:1178–1181.
e308. Dang M, Pulkkinen L, Smith FJ, McLean WH, Uitto J. Novel compound heterozygous mutations in the plectin gene in epidermolysis bullosa with muscular dystrophy and the use of protein truncation test for detection of premature termination codon mutations. Lab Invest 1998;78:195–204.
e309. Bushby KM, Gardner-Medwin D. The clinical, genetic and dystrophin characteristics of
Becker muscular dystrophy. I. Natural history. J Neurol 1993;240:98–104.
e310. Bushby KM, Gardner-Medwin D, Nicholson LV, et al. The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy. II. Correlation of phenotype with genetic and protein abnormalities. J Neurol 1993;240:105–112.
e311. Bushby KM. Genetic and clinical correlations of Xp21 muscular dystrophy. J Inherit Metab Dis 1992;15:551–564.
e312. Agretto A, Politano L, Bossone E, et al. Pulsed Doppler tissue imaging in dystrophinopathic cardiomyopathy. J Am Soc Echocardiogr 2002;15:891–899.
e313. Ammendola E, Russo V, Politano L, Santangelo L, Calabrò R. Is heart rate variability a valid parameter to predict sudden death in patients with Becker’s muscular dystrophy? Heart 2006;92:1686–1687.
e314. Annuar AA, Wong KT, Ching AS, et al. Exercise induced cramps and myoglobinuria in dystrophinopathy--a report of three Malaysian patients. Neurol Asia 2010;15:125–131.
e315. Angelini C, Fanin M, Pegoraro E, Freda MP, Cadaldini M, Martinello F. Clinicalmolecular correlation in 104 mild X-linked muscular dystrophy patients: characterization of sub-clinical phenotypes. Neuromuscul Disord 1994:4:349–358.
e316. Anthony K, Cirak S, Torelli S, et al. Dystrophin quantification and clinical correlations in
Becker muscular dystrophy: implications for clinical trials. Brain 2011;134:3547–3559.
e317. Arahata K, Hoffman EP, Kunkel LM, et al. Dystrophin diagnosis: comparison of dystrophin abnormalities by immunofluorescence and immunoblot analyses. Proc Natl Acad Sci U S A 1989;86:7154–7158.
e318. Bardoni A, Felisari G, Sironi M, et al. Loss of Dp140 regulatory sequences is associated with cognitive impairment in dystrophinopathies. Neuromuscul Disord 2000;10:194–199.
e319. Berlit P, Stegaru-Hellring B. The heart in muscular dystrophy: an electrocardiographic and ultrasound study of 20 patients. Eur Arch Psychiatry Clin Neurosci 1991;241:177–180.
e320. Carsana A, Frisso G, Tremolaterra MR, et al. Analysis of dystrophin gene deletions indicates that the hinge III region of the protein correlates with disease severity. Ann Hum Genet 2005;69:253–259.
e321. Comi GP, Prelle A, Bresolin N, et al. Clinical variability in Becker muscular dystrophy. Genetic, biochemical and immunohistochemical correlates. Brain 1994;117:1–14.
e322. Comi LI, Nigro G, Politano L, Petretta VR. The cardiomyopathy of Duchenne/Becker consultands. Int J Cardiol 1992;34:297–305.
e323. Connuck DM, Sleeper LA, Colan SD, et al; Pedatric Cardiomyopathy Registry Study Group. Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: a comparative study from the Pediatric Cardiomyopathy Registry. Am Heart J 2008;155:998–1005.
e324. de Visser M, de Voogt WG, la Rivière GV. The heart in Becker muscular dystrophy, facioscapulohumeral dystrophy, and Bethlem myopathy. Muscle Nerve 1992;15:591– 596.
e325. Finsterer J, Stöllberger C, Keller H, Slany J, Mamoli B. Cardiac involvement in patients with myotonic dystrophy, Becker’s muscular dystrophy and mitochondrial myopathy. Herz 1997;22:96–103.
e326. Gold R, Kress W, Meurers B, Meng G, Reichmann H, Müller CR. Becker muscular dystrophy: detection of unusual disease courses by combined approach to dystrophin analysis. Muscle Nerve 1992;15:214–218.
e327. de Visser M, Bakker E, Defesche JC, Bolhuis PA, van Ommen GJ. An unusual variant of
Becker muscular dystrophy. Ann Neurol 1990;27:578–581.
e328. Hoogerwaard EM, de Voogt WG, Wilde AA, et al. Evolution of cardiac abnormalities in
Becker muscular dystrophy over a 13-year period. J Neurol 1997;244:657–653.
e329. Jefferies JL, Eidem BW, Belmont JW, et al. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation 2005;112:2799–2804.
e330. Ginjaar IB, Kneppers AL, v d Meulen JD, et al. Dystrophin nonsense mutation induces different levels of exon 29 skipping and leads to variable phenotypes within one BMD family. Eur J Hum Genet 2000;8:793–796.
e331. Gospe SM Jr, Lazaro RP, Lava NS, Grootscholten PM, Scott MO, Fischbeck KH. Familial X-linked myalgia and cramps: a nonprogressive myopathy associated with a deletion in the dystrophin gene. Neurology 1989;39:1277–1280.
e332. Mavrogeni S, Papavasiliou A, Skouteli E, Magoutas A, Dangas G. Cardiovascular magnetic resonance imaging evaluation of two families with Becker muscular dystrophy. Neuromuscul Disord 2010;20:717–719.
e333. Melacini P, Fanin M, Danieli GA, et al. Cardiac involvement in Becker muscular dystrophy. J Am Coll Cardiol 1993;22:1927–1934.
e334. Melacini P, Fanin M, Danieli GA, et al. Myocardial involvement is very frequent among patients affected with subclinical Becker’s muscular dystrophy. Circulation 1996;94:3168–3175.
e335. Melo M, Lauriano V, Gentil V, et al. Becker and limb-girdle muscular dystrophies: a psychiatric and intellectual level comparative study. Am J Med Genet 1995;60:33–38.
e336. Nicholson LV, Johnson MA, Bushby KM, et al. Integrated study of 100 patients with Xp21 linked muscular dystrophy using clinical, genetic, immunochemical, and histopathological data. Part 3. Differential diagnosis and prognosis. J Med Genet 1993;30:745–751.
e337. Nigro G, Comi LI, Politano L, et al. Evaluation of the cardiomyopathy in Becker muscular dystrophy. Muscle Nerve 1995;18:283–291.
e338. Pradhan S. Valley sign in Becker muscular dystrophy and outliers of Duchenne and Becker muscular dystrophy. Neurol India 2004;52:203–205.
e339. Ramelli GP, Joncourt F, Luetschg J, Weis J, Tolnay M, Burgunder JM.. Becker muscular dystrophy with marked divergence between clinical and molecular genetic findings: case series. Swiss Med Wkly 2006;136:189–193.
e340. Saito M, Kawai H, Akaike M, Adachi K, Nishida Y, Saito S. Cardiac dysfunction with Becker muscular dystrophy. Am Heart J 1996;132:642–647.
e341. Steare SE, Dubowitz V, Benatar A. Subclinical cardiomyopathy in Becker muscular dystrophy. Br Heart J 1992;68:304–308.
e342. Stöllberger C, Finsterer J, Keller H, Mamoli B, Slany J. Progression of cardiac involvement in patients with myotonic dystrophy, Becker’s muscular dystrophy and mitochondrial myopathy during a 2-year follow-up. Cardiology 1998;90:173–179.
e343. Matsumura K, Nonaka I, Tomé FM, et al. Mild deficiency of dystrophin-associated proteins in Becker muscular dystrophy patients having in-frame deletions in the rod domain of dystrophin. Am J Hum Genet 1993;53:409–416.
e344. Sveen ML, Jeppesen TD, Hauerslev S, Køber L, Krag TO, Vissing J. Endurance training improves fitness and strength in patients with Becker muscular dystrophy. Brain 2008;131:2824–2831.
e345. Taşdemir HA, Cil E, Topaloğlu H, et al. Cardiorespiratory function in Duchenne and
Becker muscular dystrophy. Turk J Pediatr 1996;38:307–314.
e346. Muntoni F, Mateddu A, Cianchetti C, et al. Dystrophin analysis using a panel of antidystrophin antibodies in Duchenne and Becker muscular dystrophy. J Neurol Neurosurg Psychiatry 1993;56:26–31.
e347. Nakamura A, Yoshida K, Fukushima K, et al. Follow-up of three patients with a large inframe deletion of exons 45-55 in the Duchenne muscular dystrophy (DMD) gene. J Clin Neurosci 2008;15:757–763.
e348. Nicholson LV, Johnson MA, Gardner-Medwin D, Bhattacharya S, Harris JB.
Heterogeneity of dystrophin expression in patients with Duchenne and Becker muscular dystrophy. Acta Neuropathol 1990;80:239–250.
e349. Norman AM, Thomas NS, Kingston HM, Harper PS. Becker muscular dystrophy: correlation of deletion type with clinical severity. J Med Genet 1990;27:236–239.
e350. Yilmaz A, Gdynia HJ, Baccouche H, et al. Cardiac involvement in patients with Becker muscular dystrophy: new diagnostic and pathophysiological insights by a CMR approach. J Cardiovasc Magn Reson 2008;10:50.
e351. Yoshida K, Ikeda S, Nakamura A, et al. Molecular analysis of the Duchenne muscular dystrophy gene in patients with Becker muscular dystrophy presenting with dilated cardiomyopathy. Muscle Nerve 1993;16:1161–1166.
e352. Young HK, Barton BA, Waisbren S, et al. Cognitive and psychological profile of males with Becker muscular dystrophy. J Child Neurol 2008;23:155–162.
e353. Zaidman CM, Connolly AM, Malkus EC, Florence JM, Pestronk A. Quantitative ultrasound using backscatter analysis in Duchenne and Becker muscular dystrophy.
Neuromuscul Disord 2010;20:805–809.
e354. Zatz M, Rapaport D, Vainzof M, et al. Serum creatine-kinase (CK) and pyruvate-kinase (PK) activities in Duchenne (DMD) as compared with Becker (BMD) muscular dystrophy. J Neurol Sci 1991;102:190–196.
e355. Yazaki M, Yoshida K, Nakamura A, et al. Clinical characteristics of aged Becker muscular dystrophy patients with onset after 30 years. Eur Neurol 1999;42:145–149.
e356. Sahashi K, Ibi T, Suoh H, et al. Immunostaining of dystrophin and utrophin in skeletal muscle of dystrophinopathies. Intern Med 1994;33:277–283.
e357. Sánchez-Arjona MB, Rodríguez-Uranga JJ, Giles-Lima M, et al. Spanish family with myalgia and cramps syndrome. J Neurol Neurosurg Psychiatry 2005;76:286–289.
e358. Veerapandiyan A, Shashi V, Jiang YH, Gallentine WB, Schoch K, Smith EC. Pseudometabolic presentation of dystrophinopathy due to a missense mutation. Muscle Nerve 2010;42:975–979.
e359. Brabec P, Vondrácek P, Klimes D, et al. Characterization of the DMD/BMD patient population in Czech Republic and Slovakia using an innovative registry approach. Neuromuscul Disord 2009;19:250–254.
e360. Sultan A, Fayaz M. Prevalence of cardiomyopathy in Duchenne and Becker’s muscular dystrophy. J Ayub Med Coll Abbottabad 2008;20:7–13.
e361. Arahata K, Ishihara T, Kamakura K, et al. Mosaic expression of dystrophin in symptomatic carriers of Duchenne’s muscular dystrophy. N Engl J Med 1989;320:138– 142.
e362. Azofeifa J, Voit T, Hübner C, Cremer M. X-chromosome methylation in manifesting and healthy carriers of dystrophinopathies: concordance of activation ratios among first degree female relatives and skewed inactivation as cause of the affected phenotypes. Hum Genet 1995;96:167–176.
e363. Bushby KM, Goodship JA, Nicholson LV, Johnson MA, Haggerty ID, Gardner-Medwin D. Variability in clinical, genetic and protein abnormalities in manifesting carriers of Duchenne and Becker muscular dystrophy. Neuromuscul Disord 1993;3:57–64.
e364. Golla S, Agadi S, Burns DK, et al. Dystrophinopathy in girls with limb girdle muscular dystrophy phenotype. J Clin Neuromuscul Disord 2010;11:203–208.
e365. Grain L, Cortina-Borja M, Forfar C, Hilton-Jones D, Hopkin J, Burch M. Cardiac abnormalities and skeletal muscle weakness in carriers of Duchenne and Becker muscular dystrophies and controls. Neuromuscul Disord 2001;11:186–191.
e366. Hoogerwaard EM, Bakker E, Ippel PF, et al. Signs and symptoms of Duchenne muscular dystrophy and Becker muscular dystrophy among carriers in The Netherlands: a cohort study. Lancet 1999;353:2116–2119.
e367. Hoogerwaard EM, van der Wouw PA, Wilde AA, et al. Cardiac involvement in carriers of Duchenne and Becker muscular dystrophy. Neuromuscul Disord 1999;9:347–351.
e368. Pikó H, Vancsó V, Nagy B, Bán Z, Herczegfalvi A, Karcagi V. Dystrophin gene analysis in Hungarian Duchenne/Becker muscular dystrophy families - detection of carrier status in symptomatic and asymptomatic female relatives. Neuromuscul Disord 2009;19:108– 112.
e369. Politano L, Nigro V, Nigro G, et al. Development of cardiomyopathy in female carriers of
Duchenne and Becker muscular dystrophies. JAMA 1996;275:1335–1338.
e370. Soltanzadeh P, Friez MJ, Dunn D, et al. Clinical and genetic characterization of manifesting carriers of DMD mutations. Neuromuscul Disord 2010;20:499–504.
e371. Minetti C, Chang HW, Medori R, et al. Dystrophin deficiency in young girls with sporadic myopathy and normal karyotype. Neurology 1991;41:1288–1292.
e372. Seemann N, Selby K, McAdam L, et al; Canadian Pediatric Neuromuscular Group. Symptomatic dystrophinopathies in female children. Neuromuscul Disord 2011;21:172– 177.
e373. Bialer MG, McDaniel NL, Kelly TE. Progression of cardiac disease in Emery-Dreifuss muscular dystrophy. Clin Cardiol 1991;14:411–416.
e374. Bialer MG, Bruns DE, Kelly TE. Muscle enzymes and isoenzymes in Emery-Dreifuss muscular dystrophy. Clin Chem 1990;36:427–430.
e375. Buckley AE, Dean J, Mahy IR. Cardiac involvement in Emery Dreifuss muscular dystrophy: a case series. Heart 1999;82:105–108.
e376. Canki-Klain N, Récan D, Milicić D, et al. Clinical variability and molecular diagnosis in a four-generation family with X-linked Emery-Dreifuss muscular dystrophy. Croat Med J 2000;41:389–395.
e377. Carboni N, Mura M, Mercuri E, et al. Cardiac and muscle imaging findings in a family with X-linked Emery-Dreifuss muscular dystrophy. Neuromuscul Disord 2012;22:152–158.
e378. Draminska A, Kuch-Wocial A, Szulc M, et al. Echocardiographic assessment of left ventricular morphology and function in patients with Emery-Dreifuss muscular dystrophy. Int J Cardiol 2005;102:207–210.
e379. Fidziańska A, Rowińska-Marcińska K, Hausmanowa-Petrusewicz I. Coexistence of Xlinked recessive Emery-Dreifuss muscular dystrophy with inclusion body myositis-like morphology. Acta Neuropathol 2004;107:197–203.
e380. Menache CC, Brown CA, Donnelly JH, Shapiro F, Darras BT. Identification of a novel truncating mutation (S171X) in the Emerin gene in five members of a Caucasian American family with Emery-Dreifuss muscular dystrophy. Hum Mutat 2000;16:94.
e381. Karst ML, Herron KJ, Olson TM. X-linked nonsyndromic sinus node dysfunction and atrial fibrillation caused by emerin mutation. J Cardiovasc Electrophysiol 2008;19:510– 515.
e382. Mora M, Cartegni L, Di Blasi C, et al. X-linked Emery-Dreifuss muscular dystrophy can be diagnosed from skin biopsy or blood sample. Ann Neurol 1997;42:249–253.
e383. Sakata K, Shimizu M, Ino H, et al. High incidence of sudden cardiac death with conduction disturbances and atrial cardiomyopathy caused by a nonsense mutation in the STA gene. Circulation 2005;111:3352–3358.
e384. Knoblauch H, Geier C, Adams S, et al. Contractures and hypertrophic cardiomyopathy in a novel FHL1 mutation. Ann Neurol 2010;67:136–140.
e385. Quinzii CM, Vu TH, Min KC, et al. X-linked dominant scapuloperoneal myopathy is due to a mutation in the gene encoding four-and-a-half-LIM protein 1. Am J Hum Genet 2008;82:208–213.
e386. Schessl J, Columbus A, Hu Y, et al. Familial reducing body myopathy with cytoplasmic bodies and rigid spine revisited: identification of a second LIM domain mutation in FHL1. Neuropediatrics 2010;41:43–46.
e387. Schessl J, Taratuto AL, Sewry C, et al. Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1. Brain 2009;132:452–464.
e388. Schoser B, Goebel HH, Janisch I, et al. Consequences of mutations within the C terminus of the FHL1 gene. Neurology 2009;73:543–551.
e389. Shalaby S, Hayashi YK, Nonaka I, Noguchi S, Nishino I. Novel FHL1 mutations in fatal and benign reducing body myopathy. Neurology 2009;72:375–376.
e390. Chen DH, Raskind WH, Parson WW, et al. A novel mutation in FHL1 in a family with Xlinked scapuloperoneal myopathy: phenotypic spectrum and structural study of FHL1 mutations. J Neurol Sci 2010;296:22–29.
e391. Schessl J, Zou Y, McGrath MJ, et al. Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J Clin Invest 2008;118:904–912.
e392. Selcen D, Bromberg MB, Chin SS, Engel AG. Reducing bodies and myofibrillar myopathy features in FHL1 muscular dystrophy. Neurology 2011;77:1951–1959.
e393. Sarkozy A, Windpassinger C, Hudson J, et al. Phenotypic heterogeneity in British patients with a founder mutation in the FHL1 gene. Eur J Hum Genet 2011;19:1038–1044.
e394. Shalaby S, Hayashi YK, Goto K, et al. Rigid spine syndrome caused by a novel mutation in four-and-a-half LIM domain 1 gene (FHL1). Neuromuscul Disord 2008;18:959–961.
e395. Nakano S, Engel AG, Waclawik AJ, Emslie-Smith AM, Busis NA. Myofibrillar myopathy with abnormal foci of desmin positivity. I. Light and electron microscopy analysis of 10 cases. J Neuropathol Exp Neurol 1996;55:549–562.
e396. De Bleecker JL, Engel AG, Ertl BB. Myofibrillar myopathy with abnormal foci of desmin positivity. II. Immunocytochemical analysis reveals accumulation of multiple other proteins. J Neuropathol Exp Neurol 1996;55:563–577.
e397. Claeys KG, van der Ven PF, Behin A, et al. Differential involvement of sarcomeric proteins in myofibrillar myopathies: a morphological and immunohistochemical study. Acta Neuropathol 2009;117:293–307.
e398. Claeys KG, Fardeau M, Schröder R, et al. Electron microscopy in myofibrillar myopathies reveals clues to the mutated gene. Neuromuscul Disord 2008;18:656–666.
e399. Berciano J, Gallardo E, Domínguez-Perles R, et al. Autosomal-dominant distal myopathy with a myotilin S55F mutation: sorting out the phenotype. J Neurol Neurosurg Psychiatry 2008;79:205–208.
e400. Fischer D, Kley RA, Strach K, et al. Distinct muscle imaging patterns in myofibrillar myopathies. Neurology 2008;71:758–765.
e401. Foroud T, Pankratz N, Batchman AP, et al. A mutation in myotilin causes spheroid body myopathy. Neurology 2005;65:1936–1940.
e402. Hauser MA, Horrigan SK, Salmikangas P, et al. Myotilin is mutated in limb girdle muscular dystrophy 1A. Hum Mol Genet 2000;9:2141–2147.
e403. Olivé M, Goldfarb LG, Shatunov A, Fischer D, Ferrer I. Myotilinopathy: refining the clinical and myopathological phenotype. Brain 2005;128:2315–2326.
e404. Pénisson-Besnier I, Talvinen K, Dumez C, et al. Myotilinopathy in a family with late onset myopathy. Neuromuscul Disord 2006;16:427–431.
e405. Schramm N, Born C, Weckbach S, Reilich P, Walter MC, Reiser MF. Involvement patterns in myotilinopathy and desminopathy detected by a novel neuromuscular wholebody MRI protocol. Eur Radiol 2008;18:2922–2936.
e406. Olivé M, Odgerel Z, Martínez A, et al. Clinical and myopathological evaluation of early- and late-onset subtypes of myofibrillar myopathy. Neuromuscul Disord 2011;21:533–542.
e407. Gilchrist JM, Pericak-Vance M, Silverman L, Roses AD. Clinical and genetic investigation in autosomal dominant limb-girdle muscular dystrophy. Neurology 1988;38:5–9.
e408. Goldfarb LG, Park KY, Cervenáková L, et al. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet 1998;19:402–403.
e409. Arias M, Pardo J, Blanco-Arias P, et al. Distinct phenotypic features and gender-specific disease manifestations in a Spanish family with desmin L370P mutation. Neuromuscul Disord 2006;16:498–503.
e410. Bergman JE, Veenstra-Knol HE, van Essen AJ, et al. Two related Dutch families with a clinically variable presentation of cardioskeletal myopathy caused by a novel S13F mutation in the desmin gene. Eur J Med Genet 2007;50:355–366.
e411. Dalakas MC, Dagvadorj A, Goudeau B, et al. Progressive skeletal myopathy, a phenotypic variant of desmin myopathy associated with desmin mutations. Neuromuscul Disord 2003;13:252–258.
e412. Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med 2000;342:770–780.
e413. Walter MC, Reilich P, Huebner A, et al. Scapuloperoneal syndrome type Kaeser and a wide phenotypic spectrum of adult-onset, dominant myopathies are associated with the desmin mutation R350P. Brain 2007;130:1485–1496.
e414. Goudeau B, Rodrigues-Lima F, Fischer D, et al. Variable pathogenic potentials of mutations located in the desmin alpha-helical domain. Hum Mutat 2006;27:906–913.
e415. Bär H, Goudeau B, Wälde S, et al. Conspicuous involvement of desmin tail mutations in diverse cardiac and skeletal myopathies. Hum Mutat 2007;28:374–376.
e416. Dagvadorj A, Olivé M, Urtizberea JA, et al. A series of West European patients with severe cardiac and skeletal myopathy associated with a de novo R406W mutation in desmin. J Neurol 2004;251:143–149.
e417. Olivé M, Armstrong J, Miralles F, et al. Phenotypic patterns of desminopathy associated with three novel mutations in the desmin gene. Neuromuscul Disord 2007;17:443–450.
e418. Hong D, Wang Z, Zhang W, et al. A series of Chinese patients with desminopathy associated with six novel and one reported mutations in the desmin gene. Neuropathol Appl Neurobiol 2011;37:257–270.
e419. Greenberg SA, Salajegheh M, Judge DP, et al. Etiology of limb girdle muscular dystrophy 1D/1E determined by laser capture microdissection proteomics. Ann Neurol 2012;71:141–145.
e420. Vernengo L, Chourbagi O, Panuncio A, et al. Desmin myopathy with severe cardiomyopathy in a Uruguayan family due to a codon deletion in a new location within the desmin 1A rod domain. Neuromuscul Disord 2010;20:178–187.
e421. Melberg A, Oldfors A, Blomström-Lundqvist C, et al. Autosomal dominant myofibrillar myopathy with arrhythmogenic right ventricular cardiomyopathy linked to chromosome 10q. Ann Neurol 1999;46:684–692.
e422. Reilich P, Schoser B, Schramm N, et al. The p.G154S mutation of the alpha-B crystallin gene (CRYAB) causes late-onset distal myopathy. Neuromuscul Disord 2010;20:255– 259.
e423. Selcen D, Engel AG. Myofibrillar myopathy caused by novel dominant negative alpha Bcrystallin mutations. Ann Neurol 2003;54:804–810.
e424. Sacconi S, Féasson L, Antoine JC, et al. A novel CRYAB mutation resulting in multisystemic disease. Neuromuscul Disord 2012;22:66–72.
e425. Griggs R, Vihola A, Hackman P, et al. Zaspopathy in a large classic late-onset distal myopathy family. Brain 2007;130:1477–1484.
e426. Odgerel Z, Sarkozy A, Lee HS, et al. Inheritance patterns and phenotypic features of myofibrillar myopathy associated with a BAG3 mutation. Neuromuscul Disord 2010;20:438–442.
e427. Vorgerd M, van der Ven PF, Bruchertseifer V, et al. A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy. Am J Hum Genet 2005;77:297–304.
e428. Kley RA, Hellenbroich Y, van der Ven PF, et al. Clinical and morphological phenotype of the filamin myopathy: a study of 31 German patients. Brain 2007;130:3250–3264.
e429. Williams DR, Reardon K, Roberts L, et al. A new dominant distal myopathy affecting posterior leg and anterior upper limb muscles. Neurology 2005;64:1245–1254.
e430. Duff RM, Tay V, Hackman P, et al. Mutations in the N-terminal actin-binding domain of filamin C cause a distal myopathy. Am J Hum Genet 2011;88:729–740.
e431. Guergueltcheva V, Peeters K, Baets J, et al. Distal myopathy with upper limb predominance caused by filamin C haploinsufficiency. Neurology 2011;77:2105–2114.
e432. Luan X, Hong D, Zhang W, Wang Z, Yuan Y. A novel heterozygous deletion-insertion mutation (2695-2712 del/GTTTGT ins) in exon 18 of the filamin C gene causes filaminopathy in a large Chinese family. Neuromuscul Disord 2010;20:390–396.
e433. Chai Y, Bertorini TE, McGrew FA. Hereditary inclusion-body myopathy associated with cardiomyopathy: report of two siblings. Muscle Nerve 2011;43:133–136.
e434. Mitrani-Rosenbaum S, Argov Z, Blumenfeld A, Seidman CE, Seidman JG. Hereditary inclusion body myopathy maps to chromosome 9p1-q1. Hum Mol Genet 1996;5:159–163.
e435. Adler RS, Garolfalo G, Paget S, Kagen L. Muscle sonography in six patients with hereditary inclusion body myopathy. Skeletal Radiol 2008;37:43–48.
e436. Argov Z, Eisenberg I, Grabov-Nardini G, et al. Hereditary inclusion body myopathy: the Middle Eastern genetic cluster. Neurology 2003;60:1519–1523.
e437. Kim BJ, Ki CS, Kim JW, Sung DH, Choi YC, Kim SH. Mutation analysis of the GNE gene in Korean patients with distal myopathy with rimmed vacuoles. J Hum Genet 2006;51:137–140.
e438. Li H, Chen Q, Liu F, et al. Clinical and molecular genetic analysis in Chinese patients with distal myopathy with rimmed vacuoles. J Hum Genet 2011;56:335–338.
e439. Lu X, Pu C, Huang X, Liu J, Mao Y. Distal myopathy with rimmed vacuoles: clinical and muscle morphological characteristics and spectrum of GNE gene mutations in 53 Chinese patients. Neurol Res 2011;33:1025–1031.
e440. Mori-Yoshimura M, Monma K, Suzuki N, et al. Heterozygous UDP-GlcNAc 2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE gene result in a less severe GNE myopathy phenotype compared to homozygous N-acetylmannosamine kinase domain mutations. J Neurol Sci 2012;318:100–105.
e441. Argov Z, Yarom R. “Rimmed vacuole myopathy” sparing the quadriceps. A unique disorder in Iranian Jews. J Neurol Sci 1984;64:33–43.
e442. Nonaka I, Sunohara N, Ishiura S, Satoyoshi E. Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formation. J Neurol Sci 1981;51:141–155.
e443. Nonaka I, Sunohara N, Satoyoshi E, Terasawa K, Yonemoto K. Autosomal recessive distal muscular dystrophy: a comparative study with distal myopathy with rimmed vacuole formation. Ann Neurol 1985;17:51–59.
e444. Sadeh M, Gadoth N, Hadar H, Ben-David E. Vacuolar myopathy sparing the quadriceps. Brain 1993;116:217–232.
e445. Farpour F, Tehranzadeh J, Donkervoort S, et al. Radiological features of Paget disease of bone associated with VCP myopathy. Skeletal Radiol 2012;41:329–337.
e446. Kimonis VE, Kovach MJ, Waggoner B, et al. Clinical and molecular studies in a unique family with autosomal dominant limb-girdle muscular dystrophy and Paget disease of bone. Genet Med 2000;2:232–241.
e447. Kimonis VE, Mehta SG, Fulchiero EC, et al. Clinical studies in familial VCP myopathy associated with Paget disease of bone and frontotemporal dementia. Am J Med Genet A 2008;146A:745–757.
e448. Kimonis VE, Watts GD. Autosomal dominant inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Alzheimer Dis Assoc Disord 2005;19 Suppl 1:S44– S47.
e449. Guyant-Maréchal L, Laquierrière A, Duyckaerts C, et al. Valosin-containing protein gene mutations: clinical and neuropathologic features. Neurology 2006;67:644–651.
e450. Haubenberger D, Bittner RE, Rauch-Shorny S, et al. Inclusion body myopathy and Paget disease is linked to a novel mutation in the VCP gene. Neurology 2005;65:1304–1305.
e451. Miller TD, Jackson AP, Barresi R, et al. Inclusion body myopathy with Paget disease and frontotemporal dementia (IBMPFD): clinical features including sphincter disturbance in a large pedigree. J Neurol Neurosurg Psychiatry 2009;80:583–584.
e452. Kim EJ, Park YE, Kim DS, et al. Inclusion body myopathy with Paget disease of bone and frontotemporal dementia linked to VCP p.Arg155Cys in a Korean family. Arch Neurol 2011;68:787–796.
e453. Kumar KR, Needham M, Mina K, et al. Two Australian families with inclusion-body myopathy, Paget’s disease of bone and frontotemporal dementia: novel clinical and genetic findings. Neuromuscul Disord 2010;20:330–334.
e454. Stojkovic T, Hammouda el H, Richard P, et al. Clinical outcome in 19 French and Spanish patients with valosin-containing protein myopathy associated with Paget’s disease of bone and frontotemporal dementia. Neuromuscul Disord 2009;19:316–323.
e455. Viassolo V, Previtali SC, Schiatti E, et al. Inclusion body myopathy, Paget’s disease of the bone and frontotemporal dementia: recurrence of the VCP R155H mutation in an Italian family and implications for genetic counselling. Clin Genet 2008;74:54–60.
e456. Waggoner B, Kovach MJ, Winkelman M, et al. Heterogeneity in familial dominant Paget disease of bone and muscular dystrophy. Am J Med Genet 2002;108:187–191.
e457. Watts GD, Thomasova D, Ramdeen SK, et al. Novel VCP mutations in inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Clin Genet 2007;72:420–426.
e458. Shi Z, Hayashi YK, Mitsuhashi S, et al. Characterization of the Asian myopathy patients with VCP mutations. Eur J Neurol 2012;19:501–509.
e459. van der Zee J, Pirici D, Van Langenhove T, et al. Clinical heterogeneity in 3 unrelated families linked to VCP p.Arg159His. Neurology 2009;73:626–632.
e460. Watts GD. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 2004;36:377–381.
e461. Palmio J, Sandell S, Suominen T, et al. Distinct distal myopathy phenotype caused by VCP gene mutation in a Finnish family. Neuromuscul Disord 2011;21:551–555.
e462. Sunnerhagen KS, Darin N, Tajsharghi H, Oldfors A. The effects of endurance training in persons with a hereditary myosin myopathy. Acta Neurol Scand 2004;110:80–86.
e463. Tajsharghi H, Darin N, Rekabdar E, et al. Mutations and sequence variation in the human myosin heavy chain IIa gene (MYH2). Eur J Hum Genet 2005;13:617–622.
e464. Tajsharghi H, Hilton-Jones D, Raheem O, Saukkonen AM, Oldfors A, Udd B. Human disease caused by loss of fast IIa myosin heavy chain due to recessive MYH2 mutations.
Brain 2010;133:1451–1459.
e465. Tajsharghi H, Sunnerhagen KS, Darin N, Kyllerman M, Oldfors A. Induced shift in myosin heavy chain expression in myosin myopathy by endurance training. J Neurol 2004;251:179–183.
e466. Martinsson T, Oldfors A, Darin N, et al. Autosomal dominant myopathy: missense mutation (Glu-706 --> Lys) in the myosin heavy chain IIa gene. Proc Natl Acad Sci U S
A 2000;97:14614–14619.
e467. Ahlberg G, Jakobsson F, Fransson A, Moritz A, Borg K, Edström L. Distribution of muscle degeneration in Welander distal myopathy--a magnetic resonance imaging and muscle biopsy study. Neuromuscul Disord 1994;4:55–62.
e468. Borg K, Ahlberg G, Borg J, Edström L. Welander’s distal myopathy: clinical, neurophysiological and muscle biopsy observations in young and middle aged adults with early symptoms. J Neurol Neurosurg Psychiatry 1991;54:494–498.
e469. Borg K, Solders G, Borg J, Edström L, Kristensson K. Neurogenic involvement in distal myopathy (Welander). Histochemical and morphological observations on muscle and nerve biopsies. J Neurol Sci 1989;91:53–70.
e470. Borg K, Tomé FM, Edström L. Intranuclear and cytoplasmic filamentous inclusions in distal myopathy (Welander). Acta Neuropathol 1991;82:102–106.
e471. von Tell D, Somer H, Udd B, Edström L, Borg K, Ahlberg G. Welander distal myopathy outside the Swedish population: phenotype and genotype. Neuromuscul Disord 2002;12:544–547.
e472. Hackman P, Sarparanta J, Lehtinen S, et al. Welander distal myopathy is caused by a mutation in the RNA-binding protein TIA1 [published online ahead of print Dec 13, 2012]. Ann Neurol doi: 10.1002/ana.23831.
e473. Bohlega S, Abu-Amero SN, Wakil SM, et al. Mutation of the slow myosin heavy chain rod domain underlies hyaline body myopathy. Neurology 2004;62:518–521.
e474. Dubourg O, Maisonobe T, Behin A, et al. A novel MYH7 mutation occurring independently in French and Norwegian Laing distal myopathy families and de novo in one Finnish patient. J Neurol 2011;258:1157–1163.
e475. Mastaglia FL, Phillips BA, Cala LA, et al. Early onset chromosome 14-linked distal myopathy (Laing). Neuromuscul Disord 2002;12:350–357.
e476. Meredith C, Herrmann R, Parry C, et al. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause laing early-onset distal myopathy (MPD1). Am J Hum Genet 2004;75:703–708.
e477. Muelas N, Hackman P, Lugue H, et al. MYH7 gene tail mutation causing myopathic profiles beyond Laing distal myopathy. Neurology 2010;75:732–741.
e478. Laing NG, Laing BA, Meredith C, et al. Autosomal dominant distal myopathy: linkage to chromosome 14. Am J Hum Genet 1995;56:422–427.
e479. Masuzugawa S, Kuzuhara S, Narita Y, Naito Y, Taniguchi A, Ibi T. Autosomal dominant hyaline body myopathy presenting as scapuloperoneal syndrome: clinical features and muscle pathology. Neurology 1997;48:253–257.
e480. Overeem S, Schelhaas HJ, Blijham PJ, et al. Symptomatic distal myopathy with cardiomyopathy due to a MYH7 mutation. Neuromuscul Disord 2007;17:490–493.
e481. Tajsharghi H, Oldfors A, Macleod DP, Swash M. Homozygous mutation in MYH7 in myosin storage myopathy and cardiomyopathy. Neurology 2007;68:962.
e482. Tajsharghi H, Thornell LE, Lindberg C, Lindvall B, Henriksson KG, Oldfors A. Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann Neurol 2003;54:494–500.
e483. Tasca G, Ricci E, Penttilä S, et al. New phenotype and pathology features in MYH7related distal myopathy. Neuromuscul Disord 2012;22:640–647.
e484. Feit H, Silbergleit A, Schneider LB, et al. Vocal cord and pharyngeal weakness with autosomal dominant distal myopathy: clinical description and gene localization to 5q31. Am J Hum Genet 1998;63:1732–1742.
e485. Senderek J, Garvey SM, Krieger M, et al. Autosomal-dominant distal myopathy associated with a recurrent missense mutation in the gene encoding the nuclear matrix protein, matrin 3. Am J Hum Genet 2009;84:511–518.
e486. Wallgren-Pettersson C, Lehtokari VL, Kalimo H, et al. Distal myopathy caused by homozygous missense mutations in the nebulin gene. Brain 2007;130:1465–1476.
e487. Lehtokari VL, Pelin K, Herczegfalvi A, et al. Nemaline myopathy caused by mutations in the nebulin gene may present as a distal myopathy. Neuromuscul Disord 2011;21:556–562.
e488. Cirak S, von Deimling F, Sachdev S, et al. Kelch-like homologue 9 mutation is associated with an early onset autosomal dominant distal myopathy. Brain 2010;133:2123–2135.
e489. Scoto M, Cirak S, Mein R, et al. SEPN1-related myopathies: clinical course in a large cohort of patients. Neurology 2011;76:2073–2078.
e490. Hayashi YK, Matsuda C, Ogawa M, et al. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest 2009;119:2623–2633.
e491. Rajab A, Straub V, McCann LJ, et al. Fatal cardiac arrhythmia and long-QT syndrome in a new form of congenital generalized lipodystrophy with muscle rippling (CGL4) due to PTRF-CAVIN mutations. PLoS Genet 2010;6(3):e1000874.
e492. Shastry S, Delgado MR, Dirik E, Turkmen M, Agarwal AK, Garg A. Congenital generalized lipodystrophy, type 4 (CGL4) associated with myopathy due to novel PTRF mutations. Am J Med Genet A 2010;152A(9):2245–2253.
e493. Wagner KR, Fleckenstein JL, Amato AA, et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol 2008;63(5):561–571.
e494. Mendell JR, Rodino-Klapac LR, Rosales XQ, et al. Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Ann Neurol 2010;68(5):629–638.
e495. Bäckman E, Henriksson KG. Low-dose prednisolone treatment in Duchenne and Becker muscular dystrophy. Neuromuscul Disord 1995;5(3):233–241.
e496. Neumeyer AM, Cros D, McKenna-Yasek D, et al. Pilot study of myoblast transfer in the treatment of Becker muscular dystrophy. Neurology 1998;51(2):589–592.
e497. Cittadini A, Ines Comi L, Longobardi S, et al. A preliminary randomized study of growth hormone administration in Becker and Duchenne muscular dystrophies. Eur Heart J 2003;24(7):664–672.
e498. Mendell JR, Rodino-Klapac LR, Rosales-Quintero X, et al. Limb-girdle muscular dystrophy type 2D gene therapy restores alpha-sarcoglycan and associated proteins. Ann Neurol 2009;66(3):290–297.
e499. Sandin-aldehag A, Jonsson H. Evaluation of a hand training programme for patients with
Welander distal myopathy. Scand J Occup Ther 2003;10:188–192.
e500. Herson S, Hentati F, Rigolet A, et al. A phase I trial of adeno-associated virus serotype 1γ-sarcoglycan gene therapy for limb girdle muscular dystrophy type 2C. Brain 2012;135:483–492.
e501. Miller RG, Jackson CE, Kasarskis EJ, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidencebased review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2009;73(15):1218–1226.
e502. Prater SN, Banugaria SG, DeArmey SM, et al. The emerging phenotype of long-term survivors with infantile Pompe disease. Genet Med 2012;14(9):800–810.
e503. Barba-Romero MA, Barrot E, Bautista-Lorite J, et al. Clinical guidelines for late-onset Pompe disease. Rev Neurol 2012;54(8);497–507.
e504. Groh WJ. Arrhythmias in the muscular dystrophies. Heart Rhythm 2012;9(11):1890–1895.
e505. Kwon HW, Kwon BS, Kim GB, et al. The effect of enalapril and carvedilol on left ventricular dysfunction in middle childhood and adolescent patients with muscular dystrophy. Korean Circ J 2012;42(3):184–191.
e506. Bushby K, Finkel R, Birnkrant DJ, et al; DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol 2010;9(2):177–189.
e507. Allen HD, Thrush PT, Hoffman TM, Flanigan KM, Mendell JR. Cardiac management in neuromuscular diseases. Phys Med Rehabil Clin N Am 2012;23(4):855–868.
e508. Hermans MC, Pinto YM, Merkies IS, de Die-Smulders CE, Crijns HJ, Faber CG. Hereditary muscular dystrophies and the heart. Neuromuscul Disord 2010;20(8):479–492.
e509. Pane M, Messina S, Vasco G, et al. Respiratory and cardiac function in congenital muscular dystrophies with alpha dystroglycan deficiency. Neuromuscul Disord 2012;22(8):685–689.
e510. Reardon CC, Christiansen D, Barnett ED, Cabral HJ. Intrapulmonary percussive ventilation vs incentive spirometry for children with neuromuscular disease. Arch Pediatr Adolesc Med 2005;159(6):526–531.
e511. Mills B, Bach JR, Zhao C, Saporito L, Sabharwal S. Posterior spinal fusion in children with flaccid neuromuscular scoliosis: the role of noninvasive positive pressure ventilatory support. J Pediatr Orthop 2013;33(5):488–493.
e512. Bowen RE, Abel MF, Arlet V, et al. Outcome assessment in neuromuscular spinal deformity. J Pediatr Orthop 2012;32(8):792–798.
e513. Mercado E, Alman B, Wright JB. Does spinal fusion influence quality of life in neuromuscular scoliosis? Spine (Phila Pa 1976) 2007;32(19 Suppl):S120–S125.
e514. Bridges CB, Woods L, Coyne-Beasley T; Centers for Disease Control and Prevention ACIP Adult Immunization Work Group. Advisory Committee on Immunization Practices (ACIP) recommended immunization schedule for adults aged 19 years and older--United States, 2013. MMWR Surveill Summ 2013;62(Suppl 1):9–19.
e515. Miller RG, Jackson CE, Kasarskis EJ, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice Parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2009;73(15):1227–1233.
e516. Aitkens SG, McCrory MA, Kilmer DD, Bernauer EM. Moderate resistance exercise program: its effect in slowly progressive neuromuscular disease. Arch Phys Med Rehabil 1993;74(7):711–715.
e517. Kilmer DD, McCrory MA, Wright NC, Aitkens SG, Bernauer EM. The effect of a high resistance exercise program in slowly progressive neuromuscular disease. Arch Phys Med Rehabil 1994;75(5):560–563.
e518. Wright NC, Kilmer DD, McCrory MA, Aitkens, SG, Holcomb BJ, Bernauer EM. Aerobic walking in slowly progressive neuromuscular disease: effect of a 12-week program. Arch Phys Med Rehabil 1996;77(1):64–69.
Document History
Approved by the AAN Guideline Development Subcommittee on July 13, 2013; by the AAN Practice Committee on February 3, 2014; by the AANEM Board of Directors on July 10, 2014; and by the AANI Board of Directors on July 7, 2014.
This guideline was endorsed by the American Academy of Physical Medicine and Rehabilitation on April 17, 2014; by the Child Neurology Society on July 11, 2014; by the Jain Foundation on March 14, 2013; and by the Muscular Dystrophy Foundation on August 27, 2014.
Reaffirmed by the AAN in 2023.
Creation of New Guidelines, Consensus Statements, or Position Papers
AANEM members are encouraged to submit ideas for papers that can improve the understanding of the field. The AANEM will review nominated topics on the basis of the following criteria:- Members’ needs
- Prevalence of condition
- Health impact of condition for the individual and others
- Socioeconomic impact
- Extent of practice variation
- Quality of available evidence
- External constraints on practice
- Urgency for evaluation of new practice technology