Evaluation of Distal Symmetric Polyneuropathy: The Role of Laboratory and Genetic Testing
J.D. ENGLAND, MD,1 G.S. GRONSETH, MD,2 G. FRANKLIN, MD,3 G.T. CARTER, MD,4 L.J. KINSELLA, MD,5 J.A. COHEN, MD,6 A.K. ASBURY, MD,7 K. SZIGETI, MD, PHD,8 J.R. LUPSKI, MD, PHD,9 N. LATOV, MD,10 R.A. LEWIS, MD,11 P.A. LOW, MD,12 M.A. FISHER, MD,13 D. HERRMANN, MD,14 J.F. HOWARD, MD,15 G. LAURIA, MD,16 R.G. MILLER, MD,17 M. POLYDEFKIS, MD,18 A.J. SUMNER, MD19 Report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation
- Louisiana State University Health Sciences Center, Baton Rouge, Louisiana, USA
- University of Kansas, Lawrence, Kansas, USA
- University of Washington, Seattle, Washington, USA
- Providence Health System, Southwest Washington, Seattle, Washington, USA
- Tenet-Forest Park Hospital, St. Louis, Missouri, USA
- Dartmouth Hitchcock Medical Center, Lebanon, New Hampshire, USA
- University of Pennsylvania Hospital, Philadelphia, Pennsylvania, USA
- Baylor College of Medicine, Houston, Texas, USA
- Baylor College of Medicine, Houston, Texas, USA
- Weill Medical College of Cornell, Ithaca, New York, USA
- Wayne State University School of Medicine, Detroit, Michigan, USA
- Mayo Clinic, Rochester, Minnesota
- Loyola University Chicago Stritch School of Medicine, Chicago, Illinois, USA
- University of Rochester Medical Center, Rochester, New York, USA
- University of North Carolina, Chapel Hill, North Carolina, USA
- National Neurological Institute “Carlo Besta,” Milan, Italy
- California Pacific Medical Center, San Francisco, California, USA
- Johns Hopkins Medical Institute, Baltimore, Maryland, USA
- Louisiana State University Health Sciences Center, Baton Rouge, Louisiana, USA
ABSTRACT: Distal symmetric polyneuropathy (DSP) is the most common variety of neuropathy. Since the evaluation of this disorder is not standardized, the available literature was reviewed to provide evidence-based guidelines regarding
the role of laboratory and genetic tests for the assessment of DSP. A literature review using MEDLINE, EMBASE, Science Citation Index, and Current Contents was performed to identify the best evidence regarding the evaluation of polyneuropathy published
between 1980 and March 2007. Articles were classified according to a four-tiered level of evidence scheme and recommendations were based on the level of evidence. (1) Screening laboratory tests may be considered for all patients with polyneuropathy
(Level C). Those tests that provide the highest yield of abnormality are blood glucose, serum B12 with metabolites (methylmalonic acid with or without homocysteine), and serum protein immunofixation electrophoresis (Level C). If there
is no definite evidence of diabetes mellitus by routine testing of blood glucose, testing for impaired glucose tolerance may be considered in distal symmetric sensory polyneuropathy (Level C). (2) Genetic testing is established as useful for the accurate
diagnosis and classification of hereditary neuropathies (Level A). Genetic testing may be considered in patients with cryptogenic polyneuropathy who exhibit a hereditary neuropathy phenotype (Level C). Initial genetic testing should be guided by the
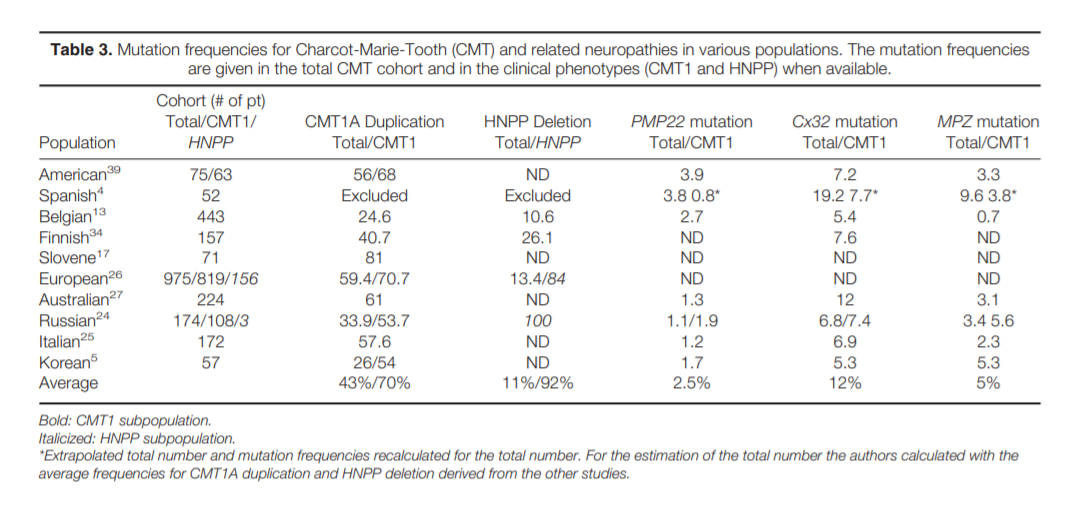
clinical phenotype, inheritance pattern, and electrodiagnostic (EDX) features and should focus on the most common abnormalities, which are CMT1A duplication/HNPP deletion, Cx32 (GJB1), and MFN2 mutation screening.
There is insufficient evidence to determine the usefulness of routine genetic testing in patients with cryptogenic polyneuropathy who do not exhibit a hereditary neuropathy phenotype (Level U).
Muscle Nerve 39: 116–125,
2009
INTRODUCTION
Justification. Polyneuropathy is a relatively common neurological disorder.8 The overall prevalence is 2,400 (2.4%) per 100,000 population, but in individuals older than 55 years the prevalence rises to 8,000 (8%) per 100,000.7,22 Since there are many etiologies of polyneuropathy; a logical clinical approach is needed for evaluation and management.This practice parameter provides recommendations for the role of laboratory and genetic tests in the evaluation of distal symmetric polyneuropathy (DSP) based on a prescribed review and analysis of the peer-reviewed literature. The parameter was developed to provide physicians with evidence-based guidelines regarding the role of laboratory and genetic tests for the assessment of polyneuropathy.
The diagnosis of DSP should be based on a combination of clinical symptoms, signs, and electrodiagnostic criteria as outlined in the previous case definition.8 [See Mission Statement, below, for details.]
Formation of Expert Panel. The Polyneuropathy Task Force included 19 physicians with representatives from the American Academy of Neurology (AAN), the American Academy of Neuromuscular and Electrodiagnostic Medicine (AANEM), and the American Academy of Physical Medicine and Rehabilitation (AAPM&R). All of the task force members had extensive experience and expertise in the area of polyneuropathy. Additionally, four members had expertise in evidence-based methodology and practice parameter development. Three are current members (J.D.E., G.S.G., G.F.), and one is a former member (R.G.M.) of the Quality Standards Subcommittee (QSS) of the AAN. The task force developed a set of clinical questions relevant to the evaluation of DSP, and subcommittees were formed to address each of these questions.
DESCRIPTION OF THE ANALYTIC PROCESS
The literature search included OVID MEDLINE (1966 to March 2007), OVID Excerpta Medica (EMBASE; 1980 to March 2007), and OVID Current Contents (2000 to March 2007). The search included articles on humans only and in all languages. The search terms
selected were peripheral neuropathy, polyneuropathy, and distal symmetric polyneuropathy. These terms were cross-referenced with the terms laboratory test, diagnosis, electrophysiology, and genetic testing.
Panel experts were asked to identify
additional articles missed by the initial search strategy. Further, the bibliographies of the selected articles were reviewed for potentially relevant articles.
Subgroups of committee members reviewed the titles and abstracts of citations
identified from the original searches and selected those that were potentially relevant to the evaluation of polyneuropathy. Articles deemed potentially relevant by any panel member were also obtained.
Each potentially relevant article
was subsequently reviewed in entirety by at least three panel members. Each reviewer graded the risk of bias in each article by using the diagnostic test classification-of-evidence scheme (Appendix 2). In this scheme, articles attaining a grade of
Class I are judged to have the lowest risk of bias, and articles attaining a grade of Class IV are judged to have the highest risk of bias. Disagreements among reviewers regarding an article’s grade were resolved through discussion. Final approval
was determined by the entire panel.
The Quality Standards Subcommittee (AAN), the Practice Issues Review Panel (AANEM), and the Practice Guidelines Committee (AAPM&R) (Appendix 1A–C) reviewed and approved a draft of the article.
The draft was next sent to members of the AAN, AANEM, and AAPM&R for further review and then to Neurology for peer review. Boards of the AAN, AANEM, and AAPM&R reviewed and approved the final version of the article. At each step of
the review process, external reviewers’ suggestions were explicitly considered. When showed that routine cerebrospinal fluid (CSF) analysis had a low diagnostic yield except in demyelinating polyneuropathies, which usually showed an increased
CSF protein level.12,28
Vitamin B12 deficiency was relatively frequent in patients with polyneuropathy, and the yield was greater when the metabolites of cobalamin (methylmalonic acid and homocysteine) were tested
(Class II and III).1,19,20,32,33 Serum methylmalonic acid and homocysteine were elevated in 5%–10% of patients whose serum B12 levels were in the low normal range of 200–500 pg/dL.20,33 In large series
of patients with polyneuropathy, between 2.2%–8% of patients had evidence of B12 deficiency as indicated by elevations of these metabolites.1,32 In one Class III study involving 27 patients with polyneuropathy and B12 deficiency,
12 (44%) had B12 deficiency based on the finding of abnormal metabolites alone.32 Thus, serum B12 assays with metabolites (methylmalonic acid and homocysteine) are useful in documenting B12 deficiency.
Although both methylmalonic acid and homocysteine are sensitive for B12 deficiency, methylmalonic acid is more specific. In a large Class III study involving 434 patients with vitamin B12 deficiency, serum methylmalonic
acid was elevated in 98.4% and serum homocysteine was elevated in 95.9%.33 In the same study serum methylmalonic acid was elevated in 12.2%, but serum homocysteine was elevated in 91% of 123 patients with isolated folate deficiency.33 Homocysteine
may also be elevated in pyridoxine deficiency and heterozygous homocysteinemia. Both homocysteine and methylmalonic acid may be elevated in hypothyroidism, renal insufficiency, and hypovolemia.
Several studies highlight the relatively high
prevalence of pre-diabetes (impaired glucose tolerance) in patients with DSP who do not fulfill the criteria for definite diabetes mellitus (Class III).30,35,37 In these studies glucose tolerance testing (GTT) was performed in patients
with idiopathic DSP. Impaired glucose tolerance was documented in 25%–36% of patients compared to 15% of controls. Additionally, patients with painful sensory polyneuropathies were more likely to have impaired glucose tolerance than those with
painless sensory polyneuropathies. Only one major study has not found an increased prevalence of impaired glucose tolerance in chronic idiopathic axonal polyneuropathy (Class III).11
Monoclonal gammopathies are more common in
patients with polyneuropathy than in the normal population. IgM monoclonal gammopathies may be associated with autoantibody activity, type I or II cryoglobulinemia, macroglobulinemia, or chronic lymphocytic leukemia. IgG or IgA monoclonal gammopathies
may be associated with myeloma, POEMS syndrome, primary amyloidosis, or chronic inflammatory conditions. In one Class III study of 279 consecutive patients with polyneuropathy of otherwise unknown etiology seen at a referral center, 10% had monoclonal
gammopathy, a significant increase over that reported in community studies.16 Serum protein immunofixation electrophoresis (IFE) is more sensitive than serum protein electrophoresis (SPEP), especially for detecting small or nonmalignant
monoclonal gammopathies. Ten of 58 (17%) monoclonal gammopathies, including 10 of 36 (30%) with IgM 5 g/L, were identified by IFE but not by SPEP.15

Conclusions.Screening laboratory tests are probably useful in determining the cause of DSP, but the yield varies depending on the particular test (Class III). The tests with the highest yield of abnormality are blood
glucose, serum B12 with metabolites (methylmalonic acid with or without homocysteine), and serum protein immunofixation electrophoresis (Class III). Patients with distal symmetric sensory polyneuropathy have a relatively high prevalence
of diabetes or pre-diabetes (impaired glucose tolerance), which can be documented by blood glucose, or GTT (Class III).
Recommendations.Screening laboratory tests may be considered for all patients with DSP (Level C).
Although routine screening with a panel of basic tests is often performed (Table 1), those tests with the highest yield of abnormality are blood glucose, serum B12 with metabolites (methymalonic acid with or without homocysteine), and serum
protein immunofixation electrophoresis (Level C). When routine blood glucose testing is not clearly abnormal, other tests for pre-diabetes (impaired glucose tolerance) such as a GTT may be considered in patients with distal symmetric sensory
polyneuropathy, especially if it is accompanied by pain (Level C).
Although there are no control studies (Level U) regarding when to recommend the use of other specific laboratory tests, clinical judgment correlated with the clinical picture
will determine which additional laboratory investigations (Table 2) are necessary.
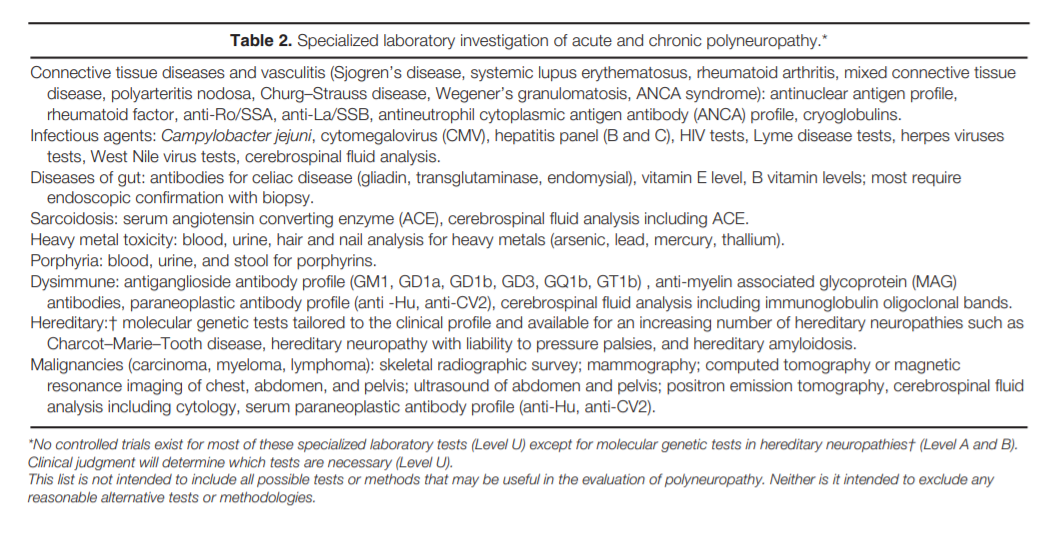
Role of Genetic Testing in the Evaluation of Polyneuropathy. Hereditary neuropathies are an important subtype of polyneuropathy, with a
prevalence of 1: 2,500 people. DSP is the predominant phenotype, but phenotypic heterogeneity may be present even within the same family; therefore, when genetic testing is contemplated all neuropathy phenotypes need to be considered.
In the evaluation of polyneuropathy a comprehensive family history should always be elicited. A high index of suspicion for a hereditary neuropathy phenotype is essential. Since molecular diagnostic techniques are available, guidelines for their usefulness
in the evaluation of polyneuropathy are needed.
|
The majority of genetically determined polyneuropathies are variants of Charcot–Marie–Tooth (CMT) disease, and genetic testing is available for an increasing number of these neuropathies. The clinical phenotype of CMT is extremely
variable, ranging from a severe polyneuropathy with respiratory failure through the classic picture with pes cavus and “stork legs” to minimal neurological findings.2,3 Since a substantial proportion of
CMT patients have de novo mutations, a family history of neuropathy may be lacking.2,3,10 Additionally, different genetic mutations can cause a similar phenotype (genetic heterogeneity) and different phenotypes can result
from the same genotype (phenotypic heterogeneity). Evaluation of Suspected Hereditary Neuropathies
RECOMMENDATIONS FOR FUTURE RESEARCHThis comprehensive review reveals several weaknesses in the current approach to the evaluation of polyneuropathy and highlights opportunities for research. |
| A= | Established as effective, ineffective, or harmful for the given condition in the specified population. (Level A rating requires as least two consistent Class I studies.) |
| B= | Probably effective, ineffective, or harmful for the givencondition in the specified population. (Level B rating requires at least one Class I study or at least two consistent Class II studies.) |
| C= | Possibly effective, ineffective, or harmful for the given condition in the specified population. (Level C rating requires at least one Class II study or two consistent Class III studies.) |
| U= | Data inadequate or conflicting; given current knowledge, treatment is unproven. |
Approved by the AANEM Board of Directors on May 1, 2008. With regard to conflicts of interest, the authors disclose the following: (1) Holds financial interests in Pfizer. (2) Holds financial interests in Pfizer and GlaxoSmithKline and Boeheringer Ingelheim for speaker honoraria and Ortho-McNeil for serving on the IDMC Committee. (3) Nothing to disclose. (4) Nothing to disclose. (5) Received royalties from the American Medical Resources, Enduring Medical Materials (CD/DVD), has received honorarium from Medical Education Resources, CME LLC, Expert Witness testimony and record review, Peters Marketing Research, Delve Marketing Research, Cross Country Education and American Medical Seminars. Dr. Kinsella holds corporate appointments with Cross Country Education and Forest Park Hospital. (6) Nothing to disclose. (7) Receives residual royalties from Elsevier for editorial work done prior to 2005. He receives honoraria from the Dana Foundation, NY, and the International Society for Neuroimmunology. His wife is a consultant for the Dana Foundation. (8) Nothing to disclose. (9) Financial interests in Athena Diagnostics and has received research funding from NIH/NEI, NIH/NIDCR, Charcot-Marie-Tooth Association, and the March of Dimes. (10) Serves as a Scientific Advisor for Quest Diagnostics and is a member of a Steering Committee, Talecris Biotherapeutics. Dr. Latov receives royalties from Demos publications and has received research support from the NIH and Talecris Biotherapeutics. He holds stock options in Therapath LLC and is the beneficiary of license fee payments from Athena Diagnostics to Columbia University. He has given expert testimony in legal proceedings related to neuropathy and has prepared an affidavit with regarding to the legal proceeding related to neuropathy. (11) Financial interests in Talecris and has received research funding from MDA and CMTA. He estimates that approximately 33% of his clinical effort is spent on electromyography. He has received payment for expert testimony regarding the use of IVIg in CIDP; neuropathic pain after breast reduction. (12) Served as a consultant for WR Medical, Viatris, Eli Lilly and Company, Chelsea Therapeutics, and Quigley Corporation. (13) Financial interests in Astrazeneca, Photothera, Wyeth, Jalmarjone Sahron, Inarx, Boehringer-Ingelheim, Dullehi-Arubio, Axaron, U-Servicer, and PAION. (14) Estimates that approximately 15%–20% of his clinical effort is spent on skin biopsies. (15) Serves on a myasthenia gravis medical scientific board, has served as an Associate Editor, Journal of Clinical Neuromuscular Disease (1998–2006), receives honoraria from Duke University Medical Center, and Medical Educational Resources. He is the director of MEG laboratories and estimates that 75% of his time is spent there. He also holds stock options in GE, Pfizer, and Johnson & Johnson. In addition, he has provided an affidavit on two cases regarding myasthenia gravis. (16) Financial interests in GlaxoSmithKline and Formenti-Grunenthal. In addition he has received research funding from Pfitzer, FormentiGrunenthal, Foramenti-Grunenthal, Italian Ministry of Health, and Regione Lombardia. (17) Financial interests in Celgene and Pathologica. (18) Financial interests in DSMB, Pfizer, Johnson & Johnson, Mitsubishi Pharma, Merck, Xenoport, and GSK. He has received research funding from JDRF, NIH, Astellas Pharma, Mitsubishi Pharma, and Sanofi-Aventis. He estimates that 10% of his clinical effort is devoted to EMG, 5% to skin biopsy, and 1% on lumbar puncture. (19) Received payment for expert testimony in the possible neurotoxic injury of the peripheral nerve.
References
[Note. Strength of evidence is indicated for references used to formulate conclusions and recommendations.]
- Barohn RJ. Approach to peripheral neuropathy and myopathy. Semin Neurol 1998;18:7–18. (Class III)
- Boerkoel CF, Takashima H, Garcia CA, et al. Charcot-MarieTooth disease and related neuropathies: mutation distribution and genotype-phenotype correlation. Ann Neurol 2002; 51:190–201. (Class I)
- Boerkoel CF, Takashima H, Lupski JR. The genetic convergence of Charcot-Marie-Tooth disease types 1 and 2 and the role of genetics in sporadic neuropathy. Curr Neurol Neurosci Rep 2002;2:70–77.
- Bort S, Nelis E, Timmerman V, et al. Mutational analysis ofthe MPZ, PMP22 and Cx32 genes in patients of Spanish ancestry with Charcot-Marie-Tooth disease and hereditary neuropathy with liability to pressure palsies. Hum Genet 1997;99:746–754. (Class I)
- Choi BO, Lee MS, Shin SH, et al. Mutational analysis ofPMP22, MPZ, GJB1, EGR2 and NEFL in Korean Charcot- Marie-Tooth neuropathy patients. Hum Mutat 2004;24:185– 186. (Class I)
- Dyck PJ, Oviatt KF, Lambert EH. Intensive evaluation of referred unclassified neuropathies yields improved diagnosis. Ann Neurol 1981;10:222–226. (Class IV)
- England JD, Asbury AK. Peripheral neuropathy. Lancet 2004; 363:2151–2161.
- England JD, Gronseth GS, Franklin G, et al. Distal symmetric polyneuropathy: a definition for clinical research. Report of the American Academy of Neurology, the American Association of Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology 2005;64:199–207.
- Fagius J. Chronic cryptogenic polyneuropathy. Acta NeurolScand 1983;67:173–180. (Class III)
- Hoogendijk JE, Hensels GW, Gabreels-Festen AA, et al. Denovo mutation in hereditary motor and sensory neuropathy type I. Lancet 1992;339:1081–1082. (Class II)
- Hughes RA, Umapathi T, Gray IA, et al. A controlled investigation of the cause of chronic idiopathic axonal polyneuropathy. Brain 2004;127:1723–1730. (Class III)
- Jann S, Beretta S, Bramerio M, Defanti CA. Prospective follow-up study of chronic polyneuropathy of undetermined cause. Muscle Nerve 2001;24:1197–1201. (Class III)
- Janssen EA, Kemp S, Hensels GW, et al. Connexin32 gene mutations in X-linked dominant Charcot-Marie-Tooth disease (CMTX1). Hum Genet 1997;99:501–505. (Class I)
- Johannsen L, Smith T, Havsager A-M, et al. Evaluation of patients with symptoms suggestive of chronic polyneuropathy. J Clin Neuromusc Dis 2001;3:47–52. (Class III)
- Kahn SN, Bina M. Sensitivity of immunofixation electrophoresis for detecting IgM paraproteins in serum. Clin Chem 1988;34:1633–1635.
- Kelly JJ, Kyle RA, O’Brien PC, Dyck PJ. Prevalence of monoclonal proteins in peripheral neuropathy. Neurology 1981;31: 1480–1483. (Class III)
- Leonardis L, Zidar J, Ekici A, Peterlin B, Rautenstrauss B.Autosomal dominant Charcot-Marie-Tooth disease type 1A and hereditary neuropathy with liability to pressure palsies: detection of the recombination in Slovene patients and exclusion of the potentially recessive Thr118MetPMP22 point mutation. Int J Mol Med 1998;1:495–501. (Class III)
- Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med 1988;318:1720–1728.
- Lindenbaum J, Rosenberg IH, Wilson PWF, Stabler SP, AllenRH. Prevalence of cobalamin deficiency in the Framingham elderly population. Am J Clin Nutr 1994;60:2–11. (Class II)
- Lindenbaum J, Savage DG, Stabler SP, et al. Diagnosis of cobalamin deficiency. II. Relative sensitivities of serum cobalamin, methylmalonic acid and total homocysteine concentrations. Am J Hematol 1990;34:99–107. (Class II)
- Lubec D, Muellbacher W, Finsterer J, Mamoli B. Diagnosticwork-up in peripheral neuropathy: an analysis of 171 cases. Postgrad Med J 1999;75:723–727. (Class III)
- Martyn CN, Hughes RAC. Epidemiology of peripheral neuropathy. J Neurol Neurosurg Psychiatry 1997;62:310–318.
- McLeod JG, Tuck RR, Pollard JD, Cameron J, Walsh JC.Chronic polyneuropathy of undetermined cause. J Neurol Neurosurg Psychiatry 1984;47:530–535. (Class III)
- Mersiyanova IV, Ismailov SM, Polyakov AV, et al. Screeningfor mutations in the peripheral myelin genes PMP22, MPZ and Cx32 (GJB1) in Russian Charcot-Marie-Tooth neuropathy patients. Hum Mutat 2000;15:340–347. (Class II)
- Mostacciuolo ML, Righetti E, Zortea M, et al. Charcot-MarieTooth disease type 1 and related demyelinating neuropathies: mutation analysis in a large cohort of Italian families. Hum Mutat 2001;18:32–41. (Class I)
- Nelis E, Van Broeckhoven C, De Jonghe P, et al. Estimation ofthe mutation frequencies in Charcot-Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur J Hum Genet 1996;4:25–33. (Class I)
- Nicholson GA. Mutation testing in Charcot-Marie-Tooth neuropathy. Ann N Y Acad Sci 1999;883:383–388. (Class II)
- Notermans NC, Wokke JH, Franssen H, et al. Chronic idiopathic polyneuropathy presenting in middle or old age: a clinical and electrophysiological study of 75 patients. J Neurol Neurosurg Psychiatry 1993;10:1066–1071. (Class III)
- Notermans NC, Wokke JH, van der Graaf Y, Franssen H, vanDijk GW, Jennekens FG. Chronic idiopathic axonal polyneuropathy: a five year follow up. J Neurol Neurosurg Psychiatry 1994;57:1525–1527. (Class III)
- Novella SP, Inzucchi SE, Goldstein JM. The frequency of undiagnosed diabetes and impaired glucose tolerance in patients with idiopathic sensory neuropathy. Muscle Nerve 2001; 24:1229–1231. (Class III)
- Periquet MI, Novak V, Collins MP, et al. Painful sensory neuropathy: prospective evaluation using skin biopsy. Neurology 1999;53:1641–1647.
- Saperstein DS, Wolfe GI, Gronseth GS, et al. Challenges in the identification of cobalamin-deficient polyneuropathy.
- Arch Neurol 2003;60:1296–1301. (Class III)
- Savage DG, Lindenbaum J, Stabler SP, Allen RH. Sensitivity of serum methylmalonic acid and total homocysteine determinations for diagnosing cobalamin and folate deficiencies. Am J Med 1994;96:239–246. (Class III)
- Silander K, Meretoja P, Juvonen V, et al. Spectrum of mutations in Finnish patients with Charcot-Marie-Tooth disease and related neuropathies. Hum Mutat 1998;12:59–68. (Class II)
- Singleton JR, Smith AG, Bromberg MB. Painful sensory polyneuropathy associated with impaired glucose tolerance. Muscle Nerve 2001;24:1225–1228. (Class IV)
- Smith AG, Singleton JR. The diagnostic yield of a standardized approach to idiopathic sensory-predominant neuropathy. Arch Intern Med 2004;164:1021–1025.
- Sumner CJ, Sheth S, Griffin JW, Cornblath DR, Polyfefkis M. The spectrum of neuropathy in diabetes and impaired glucose tolerance. Neurology 2003;60:108–111. (Class III)
- Verhoeven K, Claeys KG, Zuchner S, et al. MFN2 mutation distribution and genotype/phenotype correlation in CharcotMarie-Tooth type 2. Brain 2006;129:2093–2102. (Class II)
- Wise CA, Garcia CA, Davis SN, et al. Molecular analyses of unrelated Charcot-Marie-Tooth (CMT) disease patients suggest a high frequency of the CMTIA duplication. Am J Hum Genet 1993;53:853–863. (Class II)
- Wolfe GI, Baker NS, Amato AA, et al. Chronic cryptogenicsensory polyneuropathy: clinical and laboratory characteristics. Arch Neurol 1999;56:540–547. (Class III)
Document History
Creation of New Guidelines, Consensus Statements, or Position Papers
AANEM members are encouraged to submit ideas for papers that can improve the understanding of the field. The AANEM will review nominated topics on the basis of the following criteria:- Members’ needs
- Prevalence of condition
- Health impact of condition for the individual and others
- Socioeconomic impact
- Extent of practice variation
- Quality of available evidence
- External constraints on practice
- Urgency for evaluation of new practice technology