Consensus Criteria for the Diagnosis of Multifocal Motor Neuropathy (Consensus Statement)
ABSTRACT
At this time, there are no widely accepted criteria for the diagnosis of multifocal motor neuropathy. Furthermore, there is insufficient empirical data to define clinical and laboratory features that may reliably separate certain lower motor neuron syndromes with overlapping features as distinct. The AAEM therefore developed five criteria through a formal consensus process that are described in this document to act as a guide for diagnosing multifocal motor neuropathy with a high level of confidence (definite multifocal motor neuropathy) or with a moderate level of confidence (probable motor neuropathy). In brief, the diagnosis requires clinical weakness without objective sensory loss or upper motor neuron signs in the distribution of two or more named nerves that is due to conduction block in two or more motor nerves outside of common entrapment sites. Furthermore, normal results are required for sensory nerve conduction studies.
CONSENSUS CRITERIA FOR THE DIAGNOSIS OF MULTIFOCAL MOTOR NEUROPATHY
RICHARD K. OLNEY, MD, RICHARD A. LEWIS, MD, TIMOTHY D. PUTNAM, MD, and JOSEPH V. CAMPELLONE, JR., MD
Expert panel: Richard J. Barohn, MD; Jasper R. Daube, MD; Gerald Felsenthal, MD; Jun Kimura, MD; Richard A. Lewis, MD; Robert G. Miller, MD; Shin J. Oh, MD; Richard K. Olney, MD; Gareth J.G. Parry, MB, ChB; Austin J. Sumner, MD
Multifocal motor neuropathy is a disease of lower motor neurons in adults that produces asymmetrical muscle weakness, often in association with fasciculations and cramping. Although the weakness caused by this uncommon disease is reversible with certain immunomodulating treatments, multifocal motor neuropathy may be mistaken for other, far more serious, disorders such as amyotrophic lateral sclerosis (ALS) that do not respond to immunotherapy. At this time, there are no widely accepted criteria for the diagnosis of multifocal motor neuropathy. Furthermore, there is insufficient empirical data to define clinical and laboratory features that may reliably separate certain lower motor neuron syndromes with overlapping features as distinct. An expert panel of the American Association of Electrodiagnostic Medicine (AAEM) therefore developed this document to act as a guide for diagnosing the disease.
DESCRIPTION OF THE PROCESS
Physicians are often required to make diagnostic or therapeutic decisions for conditions in which empiric data and knowledge are incomplete or inconclusive. In such settings, the development of a consensus from an appropriate group of experts is often helpful in focusing research on critical questions and in providing interim guidance until the questions are answered empirically. Because the need for establishing consensus criteria is common, clinical health researchers have developed consensus methods over the past three decades.
A five-round modified Delphi process was used to develop these consensus criteria.4 The authors wrote an initial draft document, which was circulated for unsolicited comments from an expert panel. The expert panel was chosen from AAEM members who had been authors of articles concerned with concepts relevant to multifocal motor neuropathy and who held divergent opinions at the start of the consensus development process. Based on the comments from the expert panel, and areas of apparent agreement and disagreement, the document was revised and recirculated until consensus was reached.
DIAGNOSING MULTIFOCAL MOTOR NEUROPATHY
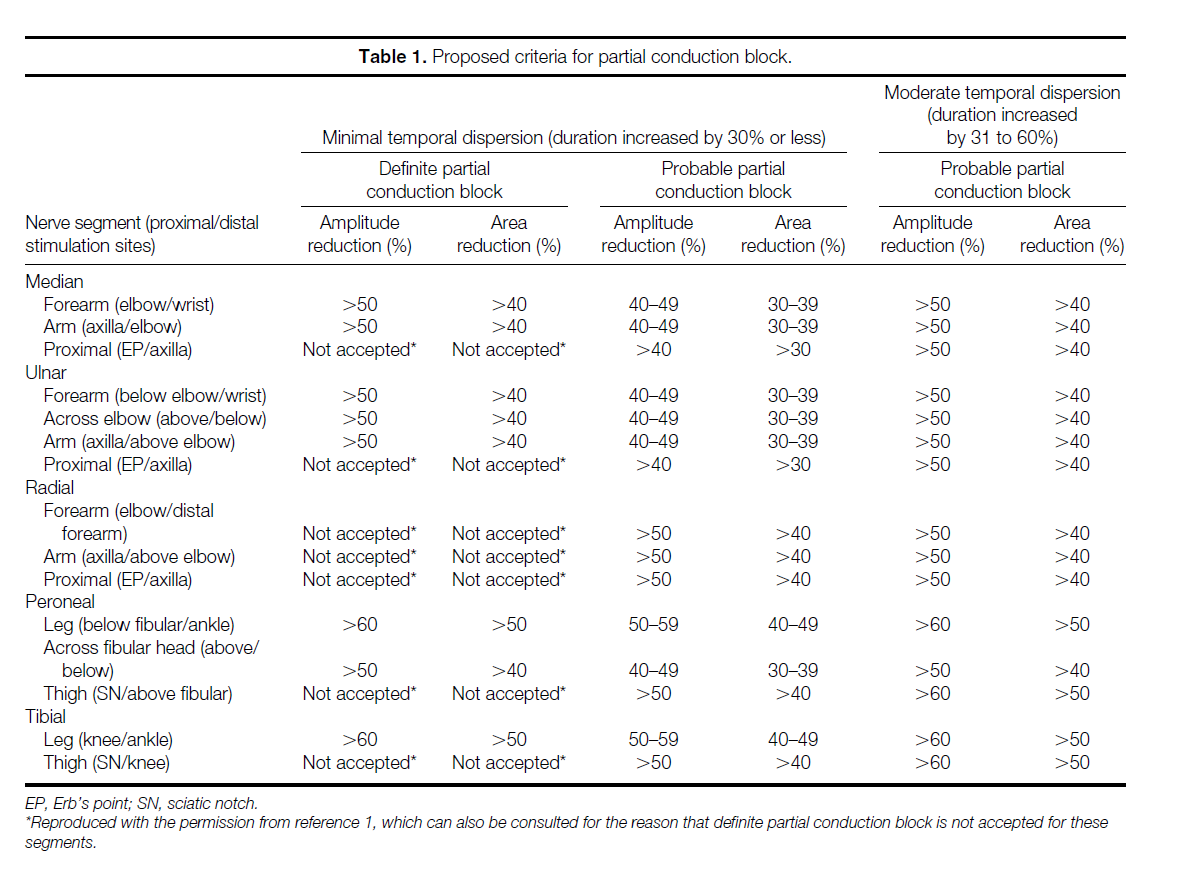
Multifocal motor neuropathy is a diagnosis that is based on recognition of a characteristic pattern of clinical symptoms, clinical signs, and electrodiagnostic findings. The fundamental electrodiagnostic finding is partial conduction block of motor axons. Thus, this consensus statement relates closely to previously described criteria for the diagnosis of partial conduction block, reproduced in brief in Table 1.1 The purpose of this consensus statement is the development of a definition for the circumstances by which multifocal motor neuropathy can be diagnosed with a high level of confidence (definite multifocal motor neuropathy) or with only a moderate level of confidence (probable multifocal motor neuropathy).
The criteria listed in Table 2 are intended to: (1) provide clinicians with diagnostic guidelines for the most typical patients, (2) propose diagnostic categories for research studies and clinical trials, and (3) stimulate further discussion about multifocal motor neuropathy. However, because these criteria are intentionally restrictive, clinicians might recommend treatment for some patients who do not satisfy these proposed criteria.
REVIEW OF THE LITERATURE ON MULTIFOCAL MOTOR NEUROPATHY
Multifocal motor neuropathy produces insidiously progressive weakness that is characteristically caused by persistent conduction block and is often associated with anti-GM1 ganglioside antibodies. Different authors first reported various aspects of the syndrome.3,8,12,13,18,19 Lewis and colleagues first described a syndrome of chronic asymmetric weakness due to persistent motor conduction block in 5 of their 40 patients with chronic acquired demyelinating polyneuropathy.8 These five patients presented with pain and numbness in one or both hands. Parry and Clarke first described patients with chronic, asymmetric weakness due to persistent motor conduction block who did not have objective sensory abnormalities.12,13 Between the time that Parry and Clarke reported their finding on the two patients in an abstract and their finding on five patients in a full paper, Roth and colleagues reported two patients with asymmetric upper limb weakness without objective sensory involvement due to persistent motor conduction block.18,19

Freddo and colleagues first recognized IgM antibody with activity directed against GM1 ganglioside in a patient with lower motor neuron weakness and a monoclonal gammopathy in 1986.3 Parry and colleagues were the first to describe and pathologically confirm a patient with a syndrome resembling motor neuron disease associated with monoclonal IgM protein that appeared to be responsive to immunosuppression.14 Pestronk and colleagues reported two patients with a reversible syndrome of motor neuron disease in which both patients presented with asymmetric hand weakness without numbness due to persistent motor conduction block that was associated with high titers of IgM anti-GM1 ganglioside antibody without a monoclonal gammopathy.16 These two patients did not respond to treatment with prednisone and plasmapheresis, but did improve with cyclophosphamide. Pestronk and colleagues coined the term multifocal motor neuropathy in this article describing the association between multifocal motor neuropathy and anti-GM1 ganglioside antibodies.16
Typical clinical and laboratory features of multifocal motor neuropathy have emerged, starting with the 1990 publication of a report that included the first large series of patients with multifocal motor neuropathy.15 In this report, two-thirds of patients were men, and two-thirds were less than the age of 45. The report described that weakness usually begins in one hand and may remain restricted to that one hand for years or may gradually spread to all four limbs. Tendon reflexes may be brisk, especially early in the course of the disease; however, spasticity, clonus, extensor plantar responses, and pseudobulbar palsy do not occur. Cranial nerve signs are rare early in the course.5 Bulbar and respiratory involvement also is rare, but if this happens, it may eventually become life threatening.9 Fasciculations and cramping are common. Sensory function is normal by clinical and electrodiagnostic examination.
Because motor neuron disease and multifocal motor neuropathy have similar clinical features, patients are sometimes diagnosed with motor neuron disease rather than multifocal motor neuropathy. However, in contrast to motor neuron disease, all patients with multifocal motor neuropathy have partial conduction block of motor fibers, and many have high titers of anti-GM1 antibodies. Although weakness in multifocal motor neuropathy is characteristically caused by partial conduction block of motor fibers, degeneration of motor axons may contribute to weakness later in the course of multifocal motor neuropathy. This results in multifocal motor neuropathy having an even greater resemblance to motor neuron disease.
In contrast to motor neuron disease, patients with multifocal motor neuropathy usually respond to treatment with intravenous immunoglobulin or cyclophosphamide.2,5 However, neither patients with multifocal motor neuropathy nor patients with motor neuron disease are expected to respond to prednisone or plasmapheresis.2,5
Confusion with regard to the diagnosis of multifocal motor neuropathy is explained, in part, by its initial recognition from two different perspectives. Cases of weakness caused by persistent conduction block were first recognized among patients with chronic demyelinating polyneuropathy.8 Many later cases were identified among patients who were initially thought to have motor neuron disease.12,13
The concept of multifocal motor neuropathy has also been confused, because different authors have applied a broad range of diagnostic criteria that vary in the (1) degree of sensory involvement, (2) requirement for partial conduction block, (3) electrophysiological criteria used for partial conduction block, and (4) requirement for anti-GM1 ganglioside antibodies. Some authors have applied broader, more inclusive criteria to diagnose multifocal motor neuropathy whereas other authors have applied narrower, more restrictive criteria. In reports by authors using the broad criteria, some cases have closely resembled multifocal motor neuropathy, except for the absence of partial conduction block.7,11 Other cases reported using the broad criteria seem typical for multifocal motor neuropathy, except for finding minor abnormalities on sensory nerve conduction studies.
The authors applying the narrower, more restrictive criteria have identified new syndromes that have lower motor neuron weakness and share features of, but are distinguished from, multifocal motor neuropathy. Three such proposed lower motor neuron syndromes are: (1) a syndrome that is associated with high titers of antiglycolipid antibodies and that has a similar absence of sensory and upper motor neuron features, but is not associated with conduction block15,17; (2) Lewis-Sumner syndrome or multifocal acquired demyelinating sensory and motor neuropathy—a syndrome that has lower motor neuron weakness associated with sensory involvement due to multifocal conduction block10,20; and (3) a syndrome that resembles multifocal motor neuropathy clinically, but has multifocal axon loss electrodiagnostically.6
Participants in this consensus process believe that multifocal motor neuropathy should be distinguished from chronic inflammatory demyelinating polyneuropathy but that sufficient empiric data and knowledge are not available at this time to determine if multifocal motor neuropathy is distinct from the other lower motor neuron syndromes described above. The expert panel had the greatest difficulty in reaching consensus on the issue of excluding the diagnosis of multifocal motor neuropathy in cases with minor abnormality on sensory nerve conduction studies.
RECOMMENDATIONS FOR FUTURE RESEARCH
A strong consensus exists in the authors and expert panel that future research concerning the pathogenesis and treatment of multifocal motor neuropathy will be advanced through a narrow consensus definition, such as the one described in this article. Depending on the results of future research, other lower motor neuron syndromes may prove to be a variant of multifocal motor neuropathy, part of a spectrum of diseases closely related to multifocal motor neuropathy, or distinct clinical entities.
In conclusion, the diagnosis of multifocal motor neuropathy is a difficult one. Until further research clarifies the issue, the criteria for definitive and probable multifocal neuropathy listed in Table 2 are proposed to serve as a guide for diagnosing this disease.
DISCLAIMER
This report is provided as an educational service of the AAEM. It is based on an assessment of current scientific and clinical information. It is not intended to include all possible methods of care of a particular clinical problem or all legitimate criteria for choosing to use a specific procedure; neither is it intended to exclude any reasonable alternative methodologies. The AAEM recognizes that specific patient care decisions are the prerogative of the patient and his/ her physician and are based on all of the circumstances involved.
References
| 1. | American Association of Electrodiagnostic Medicine. Consensus criteria for the diagnosis of partial conduction block. Muscle Nerve 1999;22:S225–S229. | |
| 2. | Feldman EL, Bromberg MB, Albers JW, Pestronk A. Immunosuppressive treatment in multifocal motor neuropathy. Ann Neurol 1991;30:397–401. | |
| 3. | Freddo L, Yu RK, Latov N, Donofrio PD, Hays AP, GreenbergHS, Albers JW, Allessi AG, Keren D. Gangliosides GM1 and GD1b are antigens for IgM M-protein in a patient with motor neuron disease. Neurology 1986;36:454–458. | |
| 4. | Jones J, Hunter D. Consensus methods for medical and healthservices research. BMJ 1995;311:376–380. | |
| 5. | Kaji R, Shibasaki H, Kimura J. Multifocal demyelinating motor neuropathy: cranial nerve involvement and immunoglobulin therapy. Neurology 1992;42:506–509. | |
| 6. | Katz J, Barohn R, Kojan S, Wolfe G, Nations S, Saperstein D,Amato A. Axonal multifocal motor neuropathy without conduction block or other features of demyelination. Neurology 2002;58:615–620. | |
| 7. | Katz J, Wolfe G, Bryan W, Jackson C, Amato A, Barohn R.Electrophysiologic findings in multifocal motor neuropathy. Neurology 1997;48:700–707. | |
| 8. | Lewis RA, Sumner AJ, Brown MJ, Asbury AK. Multifocal demyelinating neuropathy with persistent conduction block. Neurology 1982;32:958–964. | |
| 9. | Magistris M, Roth G. Motor neuropathy with multifocal persistent conduction blocks. Muscle Nerve 1992;15:1056–1057. | |
| 10. | Oh SJ, Claussen GC, Kim DS. Motor and sensory demyelinating mononeuropathy multiplex (multifocal motor and sensory demyelinating neuropathy: a separate entity or a variant of chronic inflammatory demyelinating polyneuropathy? J Periph Nerv Syst 1997;2:362–369. | |
| 11. | Pakiam A, Parry G. Multifocal motor neuropathy withoutovert conduction block. Muscle Nerve 1998;21:243–245. | |
| 12. | Parry GJ, Clarke S. Pure motor neuropathy with multifocalconduction block masquerading as motor neuron disease. Muscle Nerve 1985;8:617. | |
| 13. | Parry GJ, Clarke S. Multifocal acquired demyelinating neuropathy masquerading as motor neuron disease. Muscle Nerve 1988;11:103–107. | |
| 14. | Parry GJ, Holtz SJ, Ben-Zeev D, Drori JB. Gammopathy withproximal motor axonopathy simulating motor neuron disease. Neurology 1986;36:273–276. | |
| 15. | Pestronk A, Chaudhry V, Feldman EL, Griffin JW, CornblathDR, Denys EH, Glassberg M, Kunel RW, Olney RK, Yee WC. Lower motor neuron syndromes defined by patterns of weakness, nerve conduction abnormalities, and high titers of antiglycolipid antibodies. Ann Neurol 1990;27:316–326. | |
| 16. | Pestronk A, Cornblath DR, Ilyas AA, Baba H, Quarles RH,Griffin JW, Alderson K, Adams RN. A treatable multifocal motor neuropathy with antibodies to GM1 ganglioside. Ann Neurol 1988;24:73–78. | |
| 17. | Pestronk A, Lopate G, Kornberg AJ, Elliott JL, Blume G, YeeWC, Goodnough LT. Distal lower motor neuron syndrome with high-titer serum IgM anti-GM1 antibodies: improvement following immunotherapy with monthly plasma exchange and intravenous cyclophosphamide. Neurology 1994;44: 2027–2031. | |
| 18. | Roth G, Magistris MR. Neuropathies with prolonged conduction block, single and grouped fasciculations, localized limb myokymia. Electroencephalogr Clin Neurophysiol 1987;67: 428–438. | |
| 19. | Roth G, Rohr J, Magistris MR, Ochsner F. Motor neuropathywith proximal multifocal persistent conduction block, fasciculations and myokymia. Evolution to tetraplegia. Eur Neurol 1986;25:416–423. | |
| 20. | Saperstein DS, Amato AA, Wolfe GI, Katz JS, Nations SP,Jackson CE, Bryan WW, Burns DK, Barohn RJ. Multifocal acquired demyelinating sensory and motor neuropathy: the Lewis–Sumner syndrome. Muscle Nerve 1999;22:560–566. |
Document History
Approved by the American Association of Neuromuscular & Electrodiagnostic Medicine: June 2002.
America Association of Electrodiagnostic Medicine (AAEM) became the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM) in 2004.
Muscle Nerve 27: 117–121, 2003
Creation of New Guidelines, Consensus Statements, or Position Papers
AANEM members are encouraged to submit ideas for papers that can improve the understanding of the field. The AANEM will review nominated topics on the basis of the following criteria:- Members’ needs
- Prevalence of condition
- Health impact of condition for the individual and others
- Socioeconomic impact
- Extent of practice variation
- Quality of available evidence
- External constraints on practice
- Urgency for evaluation of new practice technology